

서 론
재료 및 방법
고추 849자원 유전체 데이터 확보 및 처리
슈퍼컴퓨터 기반 초고속 유전체 변이 탐색
초고속 유전체 변이 데이터 병합
고추 자원별 특이 SNP 탐색
결과 및 고찰
슈퍼컴퓨터 기반 유전형 분석
고추 849자원 유전형 분석 결과
고추 자원 별 특이 SNP 분석
초고속 유전형 분석을 통한 R&D 효율성 증대
적 요
서 론
다양한유전자원에 대한 정확한 특성 파악은 새로운 품종 개발 성공을 위해 필수적이다. 특히, 대규모 유전형 데이터의 확보와 분석은 유전적 다양성의 이해, 유용 형질 관련 유전자의 식별, 그리고 궁극적으로는 새로운 품종 개발을 위한 강력한 기반을 제공한다(Song et al., 2015; Zou et al., 2020). 예를 들어, 내건성, 내염성, 병해충 저항성(Ha et al., 2020; You et al., 2023) 등과 관련된 유전자들의 기능과 변이를 정확히 파악하고 이해함으로써, 목표 지향적이고 효율적인 육종이 가능해진다.
차세대 염기서열 분석(NGS) 기술의 발전으로 대량의 유전체 데이터 생산이 가능해졌으며 이는 다양한 작물의 유전체 연구에 혁명적인 변화를 가져왔다. 그러나 이렇게 방대한 양의 전장 유전체 resequencing 데이터에서 정확한 유전형을 결정하는 것은 여전히 큰 도전과제이다. 일반적인 컴퓨팅 시스템에서는 이러한 분석에 막대한 시간이 소요되어, 연구를 지연시키는 주요 병목 현상이 되고 있다. 예를 들어, 수백 개의 샘플에 대한 데이터로 부터 유전형을 분석하는 데 수개월 이상이 걸릴 수 있어, 빠른 의사결정이 필요한 육종 프로그램에서는 큰 제약이 되고 있다.
예로서, 전 세계적으로 중요한 채소 작물 중 하나로, 그 경제적, 문화적 가치가 매우 큰 고추는 단순한 식재료를 넘어 식품 산업, 의약품 개발, 화장품 제조 등 다양한 분야에서 활용되고 있으며 특히 한국을 비롯한 아시아 및 멕시코 등 중남미 국가들의 식문화에서 고추는 핵심적인 위치를 차지하고 있다. 그러나 최근 전 세계적으로 발생하고 있는 기후 변화는 고추 재배에도 심각한 위협이 되고 있다. 극단적인 기온 변화, 가뭄, 홍수 등의 이상 기후 현상은 고추의 생산성과 품질에 직접적인 영향을 미치고 있을 뿐만 아니라 기후 변화로 인한 새로운 병해충의 출현과 확산은 고추 농가에 추가적인 부담을 주고 있다. 이러한 도전에 효과적으로 대응하기 위해서 환경 스트레스에 강하고 병해충에 저항성이 있는 새로운 고추 품종의 개발이 시급해 유전체학 도구를 활용한 접근이 필요하나 유전체 크기가 3 Gbp에 달해 대량 자원의 유전형 획득부터 연구의 장벽으로 작용한다.
이러한 문제를 해결하기 위해, 본 연구팀은 농촌진흥청 슈퍼컴퓨터의 대규모 병렬 처리 능력을 기반으로 높은 정확도와 신뢰성으로 유전체 분석 분야에서 널리 사용되는 도구인 GATK (Genome Analysis Toolkit) (McKenna et al., 2010)를 채용한 초고속 유전형 분석 체계를 개발하고 고추를 모델로 삼아 849자원의 유전형 데이터를 1주 이내에 확보하였기에 그 결과를 본 논문을 통해 보고하고자 한다.
재료 및 방법
고추 849자원 유전체 데이터 확보 및 처리
고추 유전체 NGS 데이터는 미국 NCBI (Sayers et al., 2022)에 등록된 Capsicum Genome sequencing and assembly 프로젝트(PRJNA801499) (Liu et al., 2023)의 502 자원의 서열과 중국 CNCB (Bao et al., 2024)에 등록된 A pepper variome map by resequencing of a representative collection of pepper accessions 프로젝트(PRJCA004361) (Cao et al., 2022)의 347 자원의 서열을 다운로드하여 총 849자원의 데이터를 확보하였다.
종 별로는 피망, 고추와 같은 일반적인 품종이 속한 C. annuum이 400여 자원으로 가장 많았으며 하바네로와 같이 매운 품종이 포함된 C. chinense와 타바스코가 포함된 C. frutescens, 남미에서 주로 재배되며 과일 같은 향과 맛이 특징인 C. baccatum 등이 100여 자원 포함되어 있다. 기타 종으로는 안데스 산맥 지역에서 재배되는 ‘로코토’ 고추가 포함된 C. pubescens를 비롯하여 야생종으로 남미에서 주로 자생하는 C. chacoense, 갈라파고스 제도와 남미 일부 지역에서 자생하는 C. galapagoense를 비롯해 C. minutiflorum, C. cardenasii, C. eximium, C. flexuosum 및 C. rhomboideum 등이 포함되어 있다.
다운로드한 총 서열의 길이는 약 29.6 Tbp이며 Quality value 등을 포함한 FASTQ 파일의 크기는 69 TB에 이른다. NCBI에서 다운로드 한 데이터 양은 약 20.6 Tbp이며 CNCB에서 다운로드한 양은 약 8.9 Tbp이다.
데이터 QC는 Trimmomatic (Bolger et al., 2014)을 이용하여 수행하였으며 이때 trimming은 기준은 디폴트 값인 QV 30이었고 처리 후 길이가 50 bp 이하인 서열은 제거하였다. Trimming 이후 NCBI 데이터는 raw 데이터의 95.7%가 paired로 남은 반면 CNCB 데이터는 96.4%의 데이터가 남아 근소하지만 CNCB 데이터의 품질이 상대적으로 나은 것으로 나타났다(Table 1, Supplementary Table 1).
대규모 유전형 분석을 위한 참조 유전체로는 국내에서 해독된 C. annuum CM334 (Kim et al., 2014) 유전체 서열을 사용하였다. CM334는 고추의 대표적인 품종으로 해독된 유전체 서열의 총 길이는 약 2,898.3 Mbp로 염색체 수준의 고품질 조립 결과와 정교한 annotation 통한 유전자 정보를 가지고 오랜 기간 de facto 표준 유전체로 사용되고 있다.
Table 1.
NGS sequence length statistics before and after Trimmomatic processing (Unit: Gbp).
슈퍼컴퓨터 기반 초고속 유전체 변이 탐색
유전체 변이를 탐색하기 위해 본 연구팀은 높은 신뢰성으로 세계적으로 가장 많이 활용되고 있는 GATK (Genome Analysis Toolkit)와 Picard를 기반으로 파이프라인을 개발하였다. 특히, 유전체 크기가 작지 않은 고추 대량 자원의 분석 시간을 줄이기 위해 파이프라인을 슈퍼컴퓨터에서 사용할 수 있도록 최적화하였다. 구체적으로 슈퍼컴퓨터의 대규모 병렬 처리 능력을 최대한 활용하여 분석 시간을 줄이기 위해 유전형 탐색을 자원단위가 아닌 염색체 단위로 수행하도록 파이프라인을 개발하였다. 단, BWA (Li & Durbin, 2010) 단계부터 염색체 단위로 reads alignment를 수행하면 하나의 reads가 여러 개의 염색체에 매핑되는 문제가 발생할 수 있어 이를 방지하기 위해 자원 단위로 모든 염색체에 대해 BWA를 수행한 후 결과 BAM 파일을 염색체 단위로 나눈 후 이후 분석 단계를 수행하였다(Fig. 1).
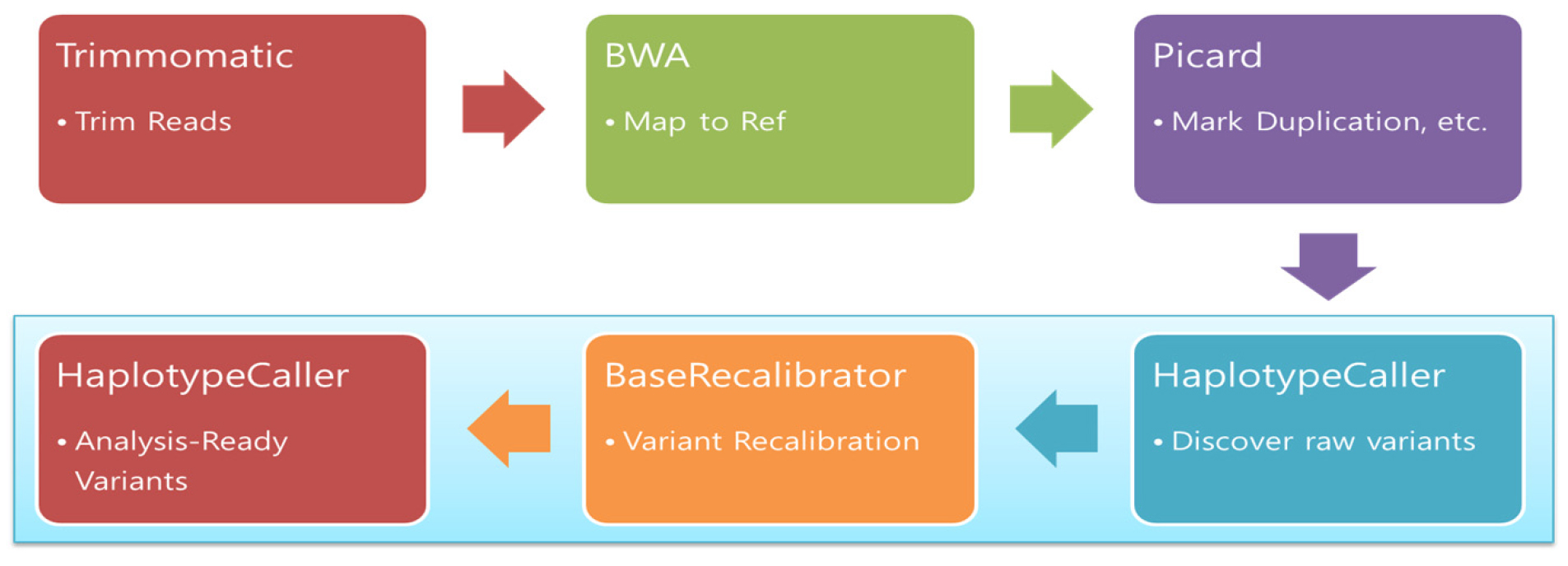
초고속 유전체 변이 데이터 병합
GATK에서 개별 자원 단위로 탐색된 변이 데이터를 다양한 분석에 활용하기 위해서는 GenotypeGVCFs로 데이터들을 모아 genotyping을 수행해 하나의 변이 파일로 전환하는 작업(joint genotyping)을 거쳐야 한다. 이를 위해 GATK에서는 GenotypeGVCFs가 변이 파일들로부터 바로 joint genotyping을 수행하는 방법과 GenomicsDBImport로 DB를 구축한 후 joint genotyping하는 두 가지 방법을 제공하고 있다. GenotypeGVCFs의 경우 상대적으로 사용이 간편하고 한 번의 처리로 결과를 얻을 수 있어 상대적으로 처리시간이 짧은 장점을 가지고 있으나 신규 자원의 데이터가 추가될 경우 작업을 처음부터 다시 수행해야 하는 단점이 있다. 반면, GenomicsDB를 이용하는 경우 변이 데이터를 DB화하는 과정과 DB로부터 joint genotyping을 하는 과정을 별도로 수행해야 하는 번거로움이 있으나 신규 자원 데이터를 추가할 경우 신규 데이터를 DB에 추가한 후 joint genotyping 과정만 수행하면 되기 때문에 보다 유연한 데이터 관리가 가능하다. 본 연구에서는 대규모 병렬 처리가 가능한 슈퍼컴퓨터의 이점을 활용하고 유연한 데이터 처리를 위해 데이터를 염색체 길이 5 Mbp 단위로 chunking하여 총 586개의 GenomicsDB를 구축하여 joint genotyping을 수행하였다.
고추 자원별 특이 SNP 탐색
고추 자원별로 특이하게 나타나는 SNP(이하 자원특이 SNP)의 탐색은 Python을 기반으로 개발한 in-house 스크립트를 이용하여 수행하였다. 스크립트는 자원별 특이 SNP의 갯수, 유전자 및 비유전자 지역에 위치하는 자원 특이 SNP 개수 및 자원특이 SNP을 보유하는 유전자의 숫자를 출력한다. 출력된 값들은 Pandas (The pandas development team, 2020), Matplotlib (Hunter, 2007) 및 Seaborn (Waskom, 2021) 등을 이용해 데이터를 분석하고 그래프를 작성하였다.
결과 및 고찰
슈퍼컴퓨터 기반 유전형 분석
고추 849자원의 유전형 분석에는 농촌진흥청 슈퍼컴퓨터에서 각 24개의 CPU 물리코어 및 128 GB의 메모리를 가진 500개의 노드가 활용되었다. 각 분석 단계별로 NGS reads의 low quality 서열 trimming 및 어댑터 서열 screening 둥 reads QC에 9시간, 표준 유전체에 대한 reads mapping 및 variant calling에 81시간 및 849자원별 valiant data를 하나로 모으는 genotype joint에 70시간 등 총 160시간(6.7일)이 소요되었다.
사용된 총 저장공간은 총 363 TB로 NCBI 및 CNCB에서 다운로드한 raw reads를 담고 있는 fastq 파일의 총 크기가 69 TB, QC를 거치며 low quality 지역 및 adapter가 제거된 reads가 56 TB, mapping 및 variant calling 결과가 가장 많은 232 TB의 저장공간을 사용하였으며 마지막으로 모든 자원의 유전형을 하나로 모으는 작업에 가장 적은 4 TB의 저장공간이 사용되었다(Table 2).
Table 2.
Data size and processing time for each stage of genotype analysis in pepper resource 849.
| Stage | Data Size | Processing Time |
| Raw FASTQ | 69 TB | - |
| FASTQ PREP | 56 TB | 9 hours |
| VARIANT CALL | 232 TB | 81 hours |
| GENOTYPE JOINT | 4 TB | 70 hours |
| TOTAL | 363 TB | 160 hours (6.7 days) |
고추 849자원 유전형 분석 결과
고추 849자원의 유전형 분석을 통해 본 연구팀은 총 625백만 개의 SNP을 식별하였다. 구체적으로 염색체 1번이 가장 많은 약 6,757만 개의 SNP를 포함하고 있는 반면 염색체 8번은 가장 적은 약 2,950만 개의 SNP를 가지고 있다(Table 3).
염색체 1 Mb 당 평균 SNP의 개수는 216,848개며 표준편차는 5,729개로 염색체 간 큰 차이는 없는 것으로 나타나 염색체별 SNP 수는 염색체 크기에 비례하였다. 고추 849자원의 SNP를 담고 있는 VCF 파일의 크기는 15 TB에 이르며 리눅스 de facto 표준 압축방식인 gzip으로 압축을 해도 1.8 TB에 달한다.
Table 3.
Number of SNPs and Variant File Size per Chromosome.
고추 자원 별 특이 SNP 분석
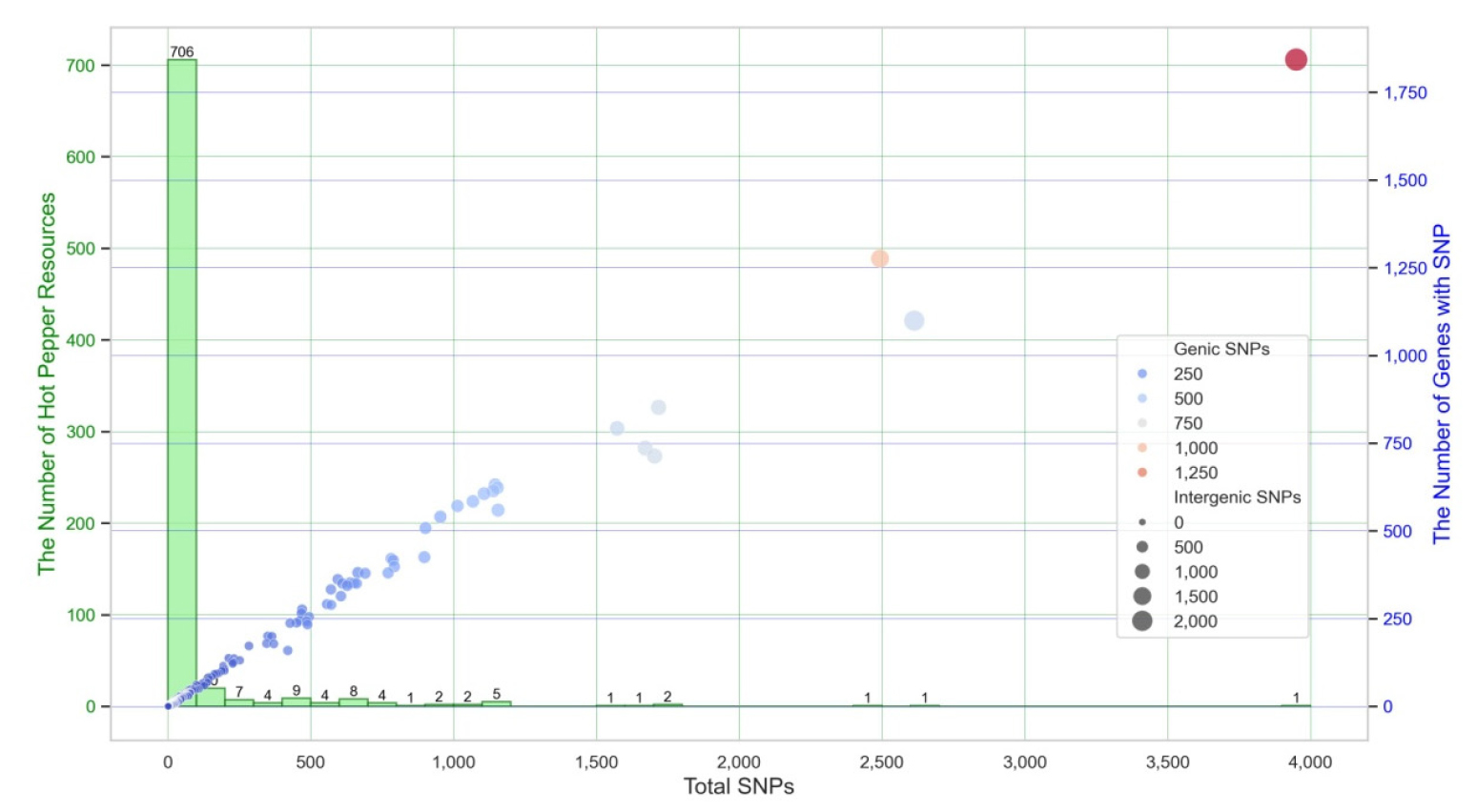
자원 별로 고유하게 가지고 있는 자원특이 SNP은 자원의 고유한 특성과 연관이 깊을 뿐만 아니라 분류 마커 개발 등 실용적인 측면에서도 매우 중요하다. 자원특이 SNP 탐색 결과 전체 849개 자원 중 779개 자원에서는 적어도 1개 이상의 자원특이 SNP을 찾을 수 있었으나 70개 자원에서는 찾을 수 없었다. 평균 개수는 73.26개이나 표준편차가 272.02개로 자원 간 편차가 크며 중간값은 8개다. 가장 많은 자원 특이 SNP을 가지 자원은 SRR19895983 (C. frutescens)로 참조유전체(C. annuum CM334) 대비 총 65.7백만 개의 SNP 중 3,950개가 자원특이 SNP이었다(Fig. 2, Supplementary Table 2).
흥미로운 것은 많은 경우 자원특이 SNP이 genic region에 더 많다는 것이다. 예를 들어 SRR19895983의 경우 전체 SNP은 당연하게도 Intergenic region에는 63.9백만 개(97.2%)가 존재하고 genic region에는 단지 1,8백만 개(2.8%)가 존재하여 유전체 내 genic region (4.9%)과 intergenic region (95.1%)의 길이 합의 비율과 유사한 반면 자원특이 SNP의 경우 intergenic region에 1,494개(37.8%)만이 위치하고 더 많은 2,456개(62.2%)의 SNP은 1,843개 유전자 지역에 위치하였다. 자원특이 SNP이 genic region에 더 많이 존재하는 현상은 여러 원인들이 복합적으로 작용한 결과로 설명할 수 있다. 구체적으로 genic region의 SNP은 유전자 기능 변경이나 발현 조절을 통해 자원의 고유 특성을 결정하고 표현형과 직접적으로 연관되어 있어 적응에 매우 중요하다. 또한, intergenic region은 무작위 변이가 쉽게 축적되는 반면, genic region은 강한 선택압으로 인해 적응적 의미가 있는 SNP만이 유지되어 자원 특이적 적응과 관련된 유의미한 변이가 집중된다(Henriques, 2018). 여기에 적응에 유리한 genic region의 변이가 선택되면서 주변 영역도 함께 고정되는 선택적 스위핑 현상도 영향을 미친다(Stephan, 2019). 이러한 진화적, 기능적 메커니즘들의 상호작용으로 인해 자원특이 SNP가 genic region에 집중되는 것으로 사료된다.
초고속 유전형 분석을 통한 R&D 효율성 증대
고추처럼 작지 않은 유전체 크기를 가진 다수 자원의 유전형 결정은 통상 오랜 분석 시간을 필요로 한다. 특히, 본 연구에서 채택한 GATK 개발 기관인 Broad 연구소에서 제안하는, 변이탐색(HaplotypeCaller)을 두 번 수행하는 방법은 결과의 신뢰성을 높일 수는 있으나 필연적으로 분석 시간을 증가시키는 원인으로 작용한다. 예를 들어 단일 작업 기준 849 자원 유전형 분석에만 총 68,769시간(81시간/자원 × 849 자원)이 필요하며 이를 많은 연구실에서 사용되는 64개의 CPU 물리코어를 가진 서버에서 각 16개의 코어를 할당하여 4개의 작업으로 수행한다고 해도 8,596 시간(716일)이 필요하다. 여기에 FASTQ 파일 QC 및 genotype joint 시간까지 감안하면 사실상 원활한 연구 진행이 불가능하다.
이러한 문제를 극복하기 위해 본 연구팀은 농촌진흥청 슈퍼컴퓨터를 기반으로 최적화된 파이프라인 개발하여 육종 프로그램과 유전체 연구에 귀중한 자원이 될 다양한 고추 자원에 대한 포괄적이고 신뢰할 수 있는 변이 데이터 셋을 1주일 이내에 확보할 수 있었다. 본 연구 성과를 통해 다양한 작물 대량 자원에 대한 유전형 분석을 고속으로 수행하여 결과를 제공할 수 있게 됨으로서 신속한 분자 마커 개발 및 유전체 선발 등 육종 효율이 향상될 것이다. 또한 빅데이터와 AI 기반의 디지털 육종 등 R&D 효율성이 크게 증대될 수 있어 궁극적으로 미래 농업에 있어서 더 나은 작물 개발과 자원 활용을 가능하게 하여 농업 경쟁력을 높이는 데 기여할 수 있을 것이다.
적 요
대규모 자원의 유전체 데이터를 확보함으로써 우리는 유전적 다양성을 더 잘 이해하고, 유용한 형질과 관련된 유전자를 식별하며, 향상된 작물 품종 개발을 위한 기반을 마련할 수 있다. 그러나 대규모 전장 유전체 resequencing 데이터의 유전형 분석에는 일반적인 컴퓨팅 시스템으로는 상당한 시간이 소요된다는 기술적 한계가 존재한다. 이에 문제 해결을 위해 우리는 RDA 슈퍼컴퓨터 시스템을 활용하여 약 3 Gbp의 유전체 크기를 가지는 849개의 고추 자원에 대한 대규모 유전체 변이 분석을 초고속으로 수행하였다. 국내에서 시퀀싱된 C. annuum CM334 참조 게놈을 기반으로 GATK 파이프라인을 통해 총 6억 2,500만 개의 SNP를 식별하였으며, 363TB의 저장 공간을 사용하여 6.7일 만에 전체 분석을 완료하였다. 분석 결과, 849개 자원 중 779개가 최소 하나 이상의 자원 특이적 SNP를 보유하고 있음을 확인하였다. 이는 대부분의 자원이 고유한 유전적 특성을 가지고 있음을 시사하며, 이러한 유전적 다양성은 향후 고추 품종 개발에 중요한 기여를 할 것으로 기대된다. 본 연구를 통해 확보된 슈퍼컴퓨팅 기반 초고속 유전형 분석 기술과 포괄적인 고추 유전체 변이 데이터셋은 각각 농촌진흥청의 농생명슈퍼컴퓨팅센터와 국가농업생명공학정보센터(NABIC)(Seol et al., 2015)를 통해 공개되어, 작물 연구 및 품종 개발을 위한 귀중한 공공 자원으로 활용될 것이다.
