

Platycodon grandiflorum, known as traditional medicine, has been extensively used since ancient times as a therapeutic to treat cold, cough and asthma (Oh et al., 2010) in Korean traditional medications. This plant is also massively used in traditional herbal medicine as a remedy for pulmonary disease and respiratory disorders (Han et al., 2000).
Platycodon grandiflorum usually called “balloon flower” or “bell flower” is well known as a perennial flowering plant that widespread in northeast Asia. The roots of this species are used as an ingredient in salads and traditional cuisine in Korea. The plant belongs to the Campanulaceae family that contains triterpenoid saponins, carbohydrates, and fibers (Tada et al., 1975). However, the annual domestic consumption of this economically important medicinal plant as a traditional cuisine in 2001 was estimated as over 4000 tons in Korea (Choi et al., 2010).
Medicinal plants are thought to be economically crops among the major and important group of crops (Rehm and Espig 1991) that have been frequently used for the prevention and treatment of many diseases and also used in the herbal medications since ancient period (Williamson, 2003). Survey conducted by World Health Organization (WHO) revealed that about 80% of the world populations still confide in the medicinal herbs. Even in the United States, the products derived from medicinal plants are used by nearly 19% of the adult populations (Kennedy 2005; Patwardhan et al., 2005).
However, previous results showed that the root of Platycodon grandiflorum (PG) reveals a wide range of pharmacologic properties, like reducing adiposity (Park et al., 2007), hyperlipidemia (Kim et al., 1995), and antiatherosclerotic effects (Zhao et al., 2006).
In other studies also reveal that components derived from Platycodon root are more effective against asthma, anti-inflammatory effects and potential against fatty liver (Ahn et al., 2005; Choi et al., 2009; Khanal et al., 2009). In addition, the most prominent component, platycosides have been manifested many biological activities, such as anti-inflammatory, antiallergy, antitumor, antiobesity, and antihyperlipidemia effects, as well as controversial immune responses and accelerating apoptosis in skin cells (Ahn et al., 2005; Kim et al., 2006; Lee et al., 2004; Wang et al., 2004; Zhao et al., 2005). Furthermore, recent investigation confirms that the main chemical component of P. grandiflorum, triterpenoidal saponins have been regarded as being responsible for these various biological effects (Choi et al., 2010).
Proteomics is thought to be a high throughput technique that has already been applied in various area of plant biology, including the study of proteins of biosynthetic pathways and its molecular function. However, in spite of a few researches in proteome investigation of some medicinal plants, the attained results so far make it explainable that proteomics still has to make good much of its presumed potential in this important area. In the past decades, two dimensional gel electrophoresis based proteomics approaches have been applied systematically to identify proteins from various medicinal plants such as Catharanthus roseus (Jacobs et al., 2005), Panax ginseng (Nam et al., 2005), Chelidonium majus (Nawrot et al., 2007), Papaver somniferum (Decker et al., 2000) and Salvia miltiorrhiza (Hung et al., 2010; Lin and Hsieh, 2010). As far goes our knowledge, research has not been accomplished yet in Platycodon grandiflorum regarding proteomics.
However, in spite of its potential medicinal significance, high throughput proteome analysis in P. grandiflorum has so far been grossly overlooked. This study reported the functional investigation from various organs of P. grandiflorum, hoping to provide references on the characterization of molecular function and characteristics for this economically important medicinal plant.
MATERIALS AND METHODS
Plant growth and preparation of P. grandiflorum extracts
Seeds of Platycodon grandiflorum were soaked on gauze, moisture with distilled water for 24 h and then the seeds were planted in vitro condition for one year. After one year, the stems were cut and sub-cultured it. Then the seedlings were raised in a controlled growth chamber at 25±1°C, 16 h photoperiod supplied by cool white fluorescent light of 1900 lux. The various plant organs; nodal section (treated as control), root, callus and shoots were collected from the sub-cultured plants. These organs were used for proteome analysis for the functional characterization and protein profiling in Platycodon grandiflorum. Three independent biological replicates were prepared for each sample.
Extraction of proteins
Total protein extract was prepared using the previously described modified protocol (Kamal, 2011). In briefly, the various organs of Platycodon grandiflorum were ground in liquid nitrogen. The organs were then suspended in solution I [(10% trichloroacetic acid (TCA) in acetone containing and 0.07% 2-mercaptoethanol (2-ME)] and then sonicate for 5-10 min. Solution II were added in the pellets and vortex, and then centrifuged at 20,000×g at 4°C for 5 min. This step was repeated and the pellets were dried by vacuum centrifugation for 10 min. Prior to incubation at room temperature for 1 hr, the dried powder was solubilized with lysis buffer (7 M urea, 2 M thiourea, 5% CHAPS, and 2 mM tributylphosphine), and then centrifuged at 20,000×g at 4°C for 20 min. The supernatants were collected to 1.5 ml tube. The protein concentrations were determined by RC/DC assay and the samples were immediately subjected to 2-DE or stored at -80°C for further utilization.
Two-dimensional gel electrophoresis and image analysis
The high throughput two-dimensional gel electrophoresis was executed according to the protocol of O’Farrell (O'Farrell 1975). Sample solutions (400 μg) were loaded onto the acidic side of the IEF tube gels (11 cm x 2 mm), which was pre-run at 150 V for 1 h, 300 V for 1 h, and 500 V for 16 h for the first dimension. SDS-PAGE in the second dimension (Nihon Eido, Tokyo, Japan) was performed with 12% separation and 5% stacking gels with 13 cm x 13 cm gel plates. Protein spots observed in the 2-DE gels were visualized by Coomassie Brilliant Blue (CBB R-250)- staining. Each biological sample was performed three times and the visualized gels were selected for image analysis in each replication. 2-DE gels were evaluated on an image scanner (HP Scanjet G4010, CA, USA; 300 dpi, 32 bits per pixel) and computer assisted image analysis was performed with Progenesis SameSpot software (Nonlinear Dynamics Ltd, Durham, NC, USA).
In-gel digestion
CBB-stained gel slices washed with 30% methanol, were destained with 10 mM (NH4) HCO3 in 50% ACN (Acetonitrile), squeezed for 10 min with 100% ACN (Acetonitrile) and dried by vacuum centrifugation. After destaining steps, the gel slices were reduced with 10 mM DTT in 100 mM (NH4)HCO3 at 56°C for 1hr and then alkylated with 55 mM Iodoacetamide (IAA) in 100 mM (NH4)HCO3 in the dark for 40 min. Then the gel slices were digested with 50 uL trypsin buffer (Promega Corporation, Madison, WI 53711-5399, USA) and incubated at 37°C for 16 hr. After digestion steps, the peptides were extracted with 50 mM ABC (Ammonium Bi-Carbonate) and this steps were repeated several times with a solution containing 0.1% formic acid in 50% ACN (Acetonitrile) until 200~250 ul. The solution containing eluted peptides was concentrated up to drying by vacuum centrifugation and the resultant extracts were confirmed by MALDI-TOF-TOF mass spectrometry.
Mass spectrometry (MALDI-TOF-TOF/MS) analysis
Identified spots from 2-DE gels were analyzed to evaluate the compatibility between protein extraction and mass spectrometry as well as to disclose the protein classes that populate each 2-DE gels. Mass spectra were acquired in an ABI 4700 Proteomics Analyzer (Applied Biosystems) using 3, 5-dimethoxy-4-hydroxycinnamic acid as matrix and the resulting data by the GPS Explorer package (Applied Biosystems). Peptides were dissolved in 0.5% (v/v) trifluoroacetic acid (TFA) and desalted with a ZipTip C18 (Millipore, Bedford, MA, USA). Those purified peptides were then eluted directly onto a MALDI plate by using an α-cyano-4-hydroxycinnamic acid (CHCA) matrix solution [10 mg per cm3 of CHCA in 0.5% (v/v) TFA + 50% (v/v) acetonitrile; 1:1]. All mass spectra were acquired in the reflection mode with 0-4000 m/z by a 4700 proteomics analyzer (Applied Bio-systems, Framingham, MA, USA). External calibration was performed using a standard peptide mixture of des-Arg bradykinin, angiotensin, Glufibrinopeptide B, adrenocorticotropic hormone (ACTH) clip 1-17, ACTH clip 18-39, and ACTH clip 7-38.
Bioinformatics
To identify the peptides, acquired MS/MS spectra were evaluated using Mascot Generic File (MGF) with an in-house licensed MASCOT search engine (Mascot v. 2.3.01, Matrix Science, London, UK) against the viridiplantae within the NCBInr database. MASCOT was used with the monoisotopic mass selected, a peptide mass tolerance of 50 ppm, and a fragment iron mass tolerance of 2 Da. The instrument setting was specified as MALDI-TOF/TOF. The carbamidomethylation of cysteines was set as a fixed modification whereas the oxidation of methionines was set as a variable modification. Trypsin was specified as the proteolytic enzyme with one potential missed cleavage. All proteins identified by high-scoring peptides were considered true matches, and at least two peptide matches. Protein hits were validated if the identification involved at least 10 top-ranking peptides with P < 0.05 and peptide scores > 34, and also selected false positive rate < 0.05. When those peptides matched multiple members of a protein family, the presented protein was selected based on the highest score and the greatest number of matching peptides.
RESULTS AND DISCUSSION
Separation of proteins by 2-DE
Protein samples extracted from nodal section (treated as control), root, callus and shoots of Platycodon grandiflorum were analyzed to characterize the protein expression using proteomic techniques (Fig. 1). The high throughput proteome technique is composed of the first dimensional of IEF and second dimension of 2D-PAGE. Two dimensional gels stained with CBB, a total of 58 differential expressed proteins (≥ 2-fold) were confirmed out of 839 protein spots and the identified protein spots were analyzed by mass spectrometry (Fig. 2).
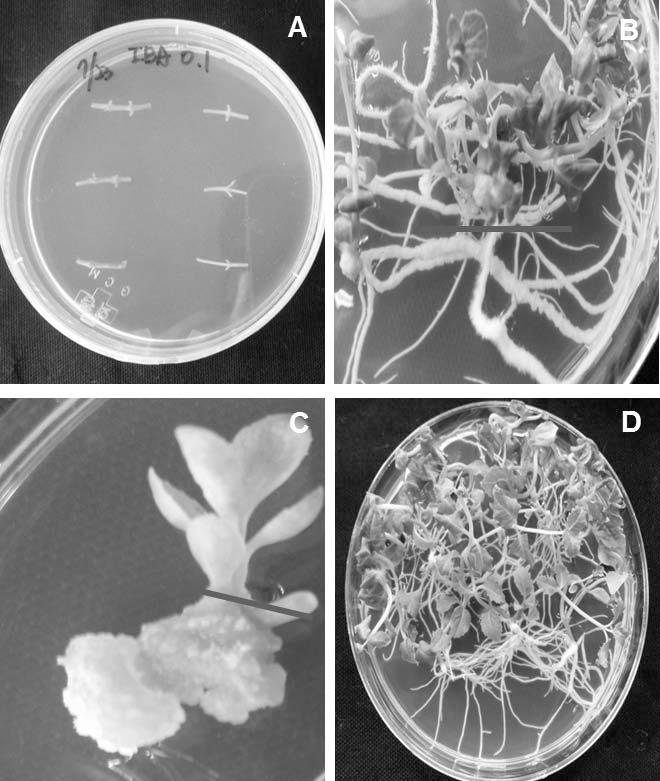
Fig. 1.
Experimental materials from different organs of Platycodon grandiflorum. A. Nodal Segment (Control), B. Root, C. Callus, D. Shoot. We used control, root, callus and shoot from 3 month’s old cultured plant for proteome analysis.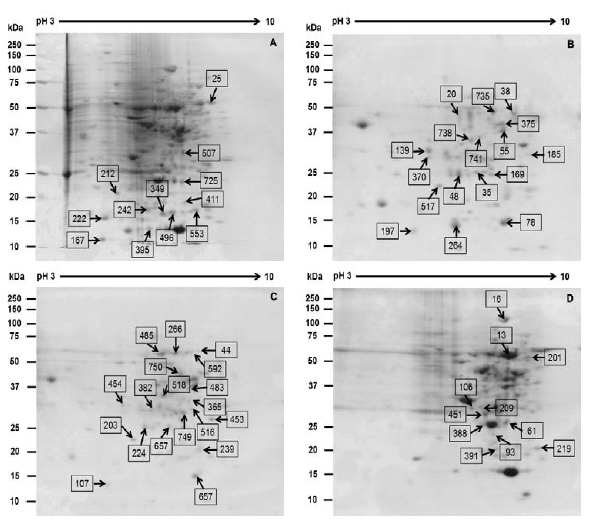
Fig. 2.
Reference 2-DE gel map from the different organs of (A) Control, (B) Callus, (C) Root, (D) Shoot. Proteins were extracted from different organs, separated by 2-DE, and visualized by CBB staining. The MW of each protein was determined by 2-DE markers. The black arrow denotes the abundance of protein increases and yellow arrow denotes decreases.Functional categorization of the identified proteins from various organs of Platycodon grandiflorum
A total of 58 proteins were classified into different possible functional categories (Molecular function, cellular component and biological process) by using iProClass database based on gene ontology. Regarding molecular function, a total of 58 differential proteins were classified into 15 functional categories whereas most of the proteins were involved in nucleic acid binding (17%), unknown (16%), transferase activity (14%), ion binding (12%), catalytic activity (7%), transporter activity (7%), isomerase activity (7%), nucleotide binding (5%), oxidoreductase activity (3%), hydrolase activity (3%) peptidase activity (2%), monooxygenase activity (2%), ligase activity (2%), chaperon (2%) and chlorophyll binding (2%) (Fig. 4(A)). In the case of cellular component, the large amounts of the identified proteins were localized in the plastid (31%), membrane (23%), nucleus (15%), mitochondrion (12%), extracellular region (8%), endoplasmic reticulum (4%), cytoplasm (4%) and ribosome (4%) respectively (Fig. 4(B)). Furthermore, considering the biological function, the identified proteins were classified into 14 categories whereas most of the proteins were involved in the metabolic process (28%), unknown (28%), oxidation-reduction process (9%), cellular protein modification process (5%), multicellular organizational process (5%), transport (5%) regulation of biological quality (3%), transcription DNA dependent (3%), proteolysis (2%), DNA replication (2%), DNA packaging (2%), syncytium formation (2%) and RNA processing (2%) respectively (Fig. 4(C)).

Identified proteins and its’ implication from various organs of Platycodon grandiflorum
Proteins, the products of the comprehensive expression of functional genes, directly reflect biological function and impair the growth and development of plants. Through a proteomic investigation of P. grandiflorum from different organs, we sought to better understand the functional categorization of individual proteins on the basis of their molecular function.
In order to identify proteins from various organs of Platycodon grandiflorum, 58 proteins were identified using MASCOT search engine according to the similarity of sequences with previously characterized proteins along with the Uniprot database (Table 1). Out of 58 protein spots, 32 protein spots were up-regulated such as ribulose-1, 5-bisphosphate carboxylase, endoplasmic oxidoreductin-1, heat stress transcription factor A3, cysteine proteinase, E3 ubiquitinprotein ligase, xyloglucan 6-xylosyltransferase, glycolate oxidase, cytochrome P450, transcription factor PCF5 respectively (Fig. 3).

Fig. 3.
Relative protein intensity of differentially expressed proteins from different organs of Platycodon grandiflorum.Table 1.
List of identified proteins from different organs of Platycodon grandiflorum using MALDI-TOF-TOF mass spectrometry.
Ribulose-1, 5-bisphosphate carboxylase acts as a key enzyme for the assimilation of carbon. In order to conserve the organic carbon, the efficiency of carbon assimilation in photosynthesis would be associated to be greatly improved and helpful. However, in the present study, this protein was regarded as the up-regulated protein (Table 1). In addition, rubisco could also bind with the important component of chlorophyll, magnesium ions; when the magnesium availability is insufficient, chlorophyll cannot be synthesized normally, and eventually, photosynthesis will be interrupted drastically (Ning et al., 2013). However, another report revealed that two spots confirmed as the small subunit of ribulose bisphosphate carboxylase (Rubisco) were decreased during seed development (Finnie et al., 2002).
Cysteine proteinase existed in multiple active isoforms in plants. In our investigation, cysteine proteinase was identified as up-regulated protein (Table 1). Though, the molecular basis and the biological relevance of these isoforms are not clear (Richau et al., 2012) and these isoforms also exposed under native conditions in Arabidopsis. However, E3 ubiquitin-protein ligase was regarded as the up-regulated protein that thought to be one of the most crucial protein which affects in the posttranslational modifications in eukaryotes (Lu et al., 2008). Previous research unlocked that the attachment of ubiquitin and ubiquitin-like polypeptides to intracellular proteins is a principal mechanism in limiting many biological processes such as proteasome degradation, intracellular trafficking, DNA repair, and signal transduction (Deshaies and Joazeiro 2009).
In proteome analysis, it is found that the effect of rapamycin on proteins related to DNA modification pathways that control DNA damage/repair and chromosomal integrity. Moreover, upregulated proteins are mostly be included substrates of the DNA damage signaling kinases, ATM kinase and ATR (53BP1, FAM44a, and MDC1) and other proteins are involved in DNA damage responses such as tankyrase 1-binding protein 1 (TNKS1BP1), NPM, and NUP98 (Bandhakavi et al., 2010). In the present investigation, putative DNA repair protein was identified as up-regulated protein (Table 1).
S-adenosylmethionine synthase 3 identified as up-regulated protein that catalyzes the formation of S-adenosylmethionine from methionine and ATP. The overall synthetic reaction is composed of two sequential steps, AdoMet formation and the subsequent tripolyphosphate hydrolysis which occurs prior to release of AdoMet from the enzyme (http://www.uniprot.org/ uniprot/P43282). In the present investigation, glycolate oxidase (GLO) was also identified as the up-regulated protein (Table 1). GLO considered as the key enzyme that involved in the photorespiration and catalyzed the oxidation of glycolate to glyoxylate, with an equimolar amount of H2O2 produced (Foyer et al., 2009). Moreover, more than 70% of the total H2O2 production in photosynthetic leaves of C3 plants derived from photorespiration via GLO catalysis (Noctor et al., 2002). Many studies also have suggested that GLO may also play efficient roles in plant stress responses. It has been investigated that the activities of GLO were induced in response to various environmental stresses which was found in vigna, pea and tobacco (Mittler and Zilinskas 1994; Mukherjee and Choudhuri 1983; Rizhsky et al., 2002).
Transcription factor PCF5 was identified as up-regulated protein which is well known for activating transcription. Transcription factor binds the promoter core sequence 5'-GGNCC-3' (Kosugi and Ohashi 2002). Furthermore, peptidyl-prolyl cis-trans isomerase CYP19-2 was also identified as up-regulated protein in the present research that catalyzes the cis-trans isomerization of proline imidic peptide bonds in oligopeptides (http://www.uniprot. org/uniprot/P43282).
However, out of 58 protein spots, a total of 26 protein spots were identified as down-regulated such as RNA recognition motif-containing protein, 60S acidic ribosomal protein P0, malate dehydrogenase, ATP synthase subunit delta mitochondrial, heat shock 22 kDa protein, putative DNA repair protein RAD 23-3, S-adenosylmethionine synthase 3, xyloglucan 6-xylosyltransferase respectively (Fig. 3).
In the present investigation, the RNA recognition motif-containing protein was identified as down-regulated protein (Table 1). Basically, RRM proteins have a wide range of RNA binding preferences and functions such as heterogeneous nuclear ribonucleoproteins (hnRNPs), regulation of alternative splicing proteins (SR, U2AF2, Sxl), small nuclear ribonucleoproteins (U1 and U2 snRNPs), and RNA stability and translation inhibiting proteins (PABP, La, Hu) (Sachs et al., 1987).
60S acidic ribosomal protein P0 is a protein that prevails in humans which is encoded by the RPLP0 gene. This protein was identified as the down-regulated protein in our investigation. In addition, this gene encodes a ribosomal protein which is a component of the 60S subunit. However, the protein is typical for genes encoding ribosomal proteins; and there are many pseudogenes of this gene scattered through the genome (Kenmochi et al., 1998).
Malate dehydrogenase (MDH) acts as an enzyme that catalyzes the oxidation of malate to oxaloacetate reversibly using the reduction of NAD+ to NADH. MDH was identified as downregulated protein. The hydrophobic cavity regarded as the active site of MDH within the protein complex has specific binding sites for the substrate and its coenzyme, NAD+. Previous study suggests that MDH endures a conformational change in its active state that efficiently encloses the substrate to minimize solvent exposure and to position key residues in closer proximity to the substrate, (Goward and Nicholls 1994).
ATP synthase subunit delta mitochondrial was identified in the present investigation that was regarded as down-regulated protein (Table 1). Mitochondrial ATP synthase is essential for the production of ATP; even the reports about the functional role of ATP are insufficient. However, structurally ATP synthase comprising of a membrane- bound moiety (F0) and a protruding globular and soluble moiety (F1) that can be released (Sawicki and Jugdutt 2007).
Xyloglucan 6-xylosyltransferase was identified in the present research that characterized as down-regulated protein (Table 1). However, XXT acts as a prominent role in transferring one xylose mainly to the second glucose residue from the non-reducing end. The acceptor should have a minimum of four glucose residues (Faik et al., 2002). Therefore, it is investigated that a membrane complex of XyG glucan synthase and XTs would be more efficient than solubilized enzymes at generating the observed XXXG repeating structure in planta (Cavalier et al., 2008).
CONCLUSION
The protein profile of Platycodon grandiflorum was improved by using high throughput two-dimensional IEF-2D-PAGE together with MALDI-TOF-TOF mass spectrometry. Two dimensional gels stained with CBB, we confirmed a total of 58 differential expressed protein spots (≥ 2-fold) that were analyzed by mass spectrometry. However, the proteins were mostly involved in the nucleic acid binding, transferase activity, ion binding, catalytic activity, transporter activity, isomerase activity. Although this study was an initial proteomic investigation from the various organs of P. grandiflorum so far, it is our belief that the proteome profiling provides not only insights into medicinal plant research but also a good starting point for further investigation of the functions of these proteins using genetic and other approaches. More extensive studies are needed to fully understand the mechanism of various organs underlying the medicinal plant, P. grandiflorum.
