

옥수수(Zea mays L.)는 광합성 능력이 높은 C4 작물이기 때문에 단위 면적당 생산량이 높고 재배가 용이하여 밀, 벼 와 함께 세계 3대 중요 작물(major crop) 중 하나이다(Varshney et al., 2012). 옥수수는 전분을 원료로 하는 각종 산업의 가 장 중요한 작물 중 하나로서 식량과 사료 외에도 의약품, 시 약, 화장품, 도료, 인쇄, 제지업 등 산업 및 공업원료로서 매 우 중요한 위치를 차지하고 있다(Kim et al., 2002; Prasanna et al., 2010). 최근에는 전분을 이용한 바이오에탄올과 목질 계를 이용한 바이오에탄올 연구에도 옥수수가 다양하게 활 용되고 있다.
옥수수의 연간 총 생산량은 전 세계적으로 872,006,700 톤으로서, 지난 60년 동안 다른 식량작물이나 유지작물 (oilseed crop)에서는 유래가 없는 6배의 생산량의 증가를 보이고 있으며, 아직도 미국을 중심으로 생산량의 증가세를 보이고 있다(Lee & Tollenaar, 2007; FAOSTAT, 2012). 반 면 우리나라의 연간 옥수수 생산량은 불과 83,210톤으로서 일본, 멕시코에 이어 세계 3대 옥수수 수입국(7,758,658 톤) 인 실정이다(FAOSTAT, 2012). 우리나라에서 소비되는 대 부분의 옥수수는 거의 전량 수입에 의존하고 있는 반면 옥 수수의 육종은 국내 타 작물과 비교하여 상당히 부진한 상 황이다. 그 동안의 옥수수 육종 방법은 집단선발에 의한 재 래식 육종을 통한 방임수분 품종이 재배되었으나, 교잡종 이론이 등장하면서 다양한 교잡종 옥수수 연구를 통하여 복 교잡종 품종이 개발되었으며, 근래에는 잡종강세가 우수한 단교잡종 품종이 개발되어 재배되기 시작하였다(Bernardo, 1996; Troyer, 1999; Park et al., 2009). 최근에는 단교잡종 을 중심으로 상반 반복 선발 등을 이용한 우수한 자식 계통 개발이나 유전적 다형성을 위한 생식세포향상법(germplasm enhancement)에 대한 관심이 높아지고 있으며, 옥수수 육종 연구를 통해 수집된 옥수수 유전자원들의 유전적 다양성이 나 계통 간의 유연관계는 작물 유전체학의 발달과 더불어 새로운 신품종 개발에 이용될 것이다(Lee & Tracy, 2009).
분자육종이란 식물 육종에 분자생물학적 기술을 적용시 키는 것으로서 분자 마커를 이용하여 작물 선발에 이용하는 MAS와 QTL mapping, 유전공학, 식물 형질 전환 등의 기 술을 의미한다. 본격적인 분자 유전학 기법을 이용한 분자육 종 시대는 1980년대 초반 Agrobacterium을 이용한 형질전환 체가 처음 개발 되면서 시작되었다(Fraley et al., 1983; Herrera- Estrella et al., 1983). 그 후 생명공학 기술이 작물 육종과 개발에 활용 가능성이 확인 되면서 형질전환 작물의 상업화 에 성공하게 되었다(Koziel et al., 1993). 또한 분자 마커도 육종에 이용될 높은 가능성을 지니고 있다. 분자 마커가 개 발되기 전에는 표현형을 이용한 표지 인자를 이용하여 선발 이 이루어졌으나 이후 종자 저장 단백질과 같은 생화학적 동위 효소를 이용한 마커가 개발 되었다. 하지만 이 방법은 육종에 활용할 수 있는 시기가 한정적이기 때문에 많이 활 성화 되지 못했다(Tanksley et al., 1981; Seo et al., 1995). RFLP(Restriction Fragment Length Polymorphism) 기술의 개발은 분자 마커의 새로운 장을 열었으며, 이후 작물에 적 용되어 전형적인 분자육종의 새로운 연구 분야를 개척했다. RFLP 마커 개발 이후에는 기존의 마커와는 비교가 되지 않 을 정도로 많은 대립 유전자를 분석 할 수 있게 되었다 (Botstein et al., 1980; Helentjaris et al. 1986). 또한, DNA를 이용한 분자 마커를 이용한 표지인자 시스템이 개발되면서 정교한 수준의 유전자 지도와 분자마커와 농업적으로 중요 한 작물의 형질간의 유전적 연관을 활용한 연구도 진행되었 으며, 특히 옥수수 육종에서 이용될 잠재력을 인정받게 되었 다(Edwards et al., 1987; Paterson et al., 1988). 오늘날에는 분자생물학 기술의 발전으로 RAPD(Random Amplified Polymorphic DNA), AFLP(Amplified Fragment Length Polymorphism) 및 SSR(Simple Sequence Repeat)등의 다양 한 DNA 분석기법들이 개발되어 있다. 분자마커 기술은 타 식성 작물인 옥수수에 다양한 유전적 변이성과 각 계통 간 의 유연관계 등에 대한 정보를 제공 할 수 있기 때문에 신 품종의 개량이나 자식 계통의 육성 및 발굴, 그리고 교배 조 합의 예측 등 다양하게 유용한 육종 프로그램에 이용될 수 있다(Park et al., 2009; Sa et al., 2011). 본 고에서는 최근 Next Generation Sequencing 시대를 맞아 폭발적으로 성장 하고 있는 분자마커를 옥수수 육종에 이용한 활용과 현황을 알아보고자 한다.
분자마커의 종류
Botstein et al.(1980)에 의해 처음 RFLP 기술을 genetic mapping에 이용하여 분자마커 기술로 보고한 이후에 여러 가지 분자마커들이 개발되었다. 일반적으로 분자마커 기술 은 PCR(Polymerase Chain Reaction) 기반 방법과 비 PCR 기반 방법으로 나누어서 구분한다.
비 PCR 기반(non-PCR-based) 방법
RFLP
RFLP는 제한효소에 의해 잘라진 DNA의 Southern blot 에 방사선 동위 원소로 표지된 탐침(probe)을 이용하여 DNA 의 다형성을 찾아내는 기술이다. RFLP를 이용하여 서로 다 른 DNA의 프로파일을 분석할 수 있는데, 이 프로파일의 차 이는 단일 염기 서열의 변화로 인한 염기치환 돌연변이나 유전자의 삽입, 결실, 역위 등에 인한 DNA 재배열에 의해 발생하였음을 의미 한다. RFLP 마커는 상대적으로 높은 다 형성과 공우성을 보이며 높은 재현성을 보이는 신빙성 있는 기법이나 많은 양의 DNA가 필요하고, 시간이 많이 필요하 다. 이러한 제약으로 인하여 PCR 기반 분자마커가 개발되 게 되었다.
PCR 기반(PCR-based) 방법
PCR 기법이 처음 개발된 이후에 다양한 PCR 기반 분자 마커들이 개발 되었다. 그 이유는 PCR 의 간편성과 높은 성공 가능성 때문이라고 할 수 있겠다.
임의의 프라이머(arbitrarily primer)를 이용한 PCR 기반 마커
RAPD
RAPD는 임의의 염기 서열의 단일 프라이머를 이용하여 염기서열의 다형성을 추적하는 기술로서 1가지 종류의 프 라이머가 genomic DNA 의 서로 다른 위치의 상보적 염기 서열에 결합을 하여 증폭된 DNA 다형성을 분석하는 기술 이다(Williams et al., 1990). RAPD의 장점은 적은 양의 DNA로 분석이 가능하다는 점이며 또한 빠르고, Southern blot의 과정이 필요하지 않다는 편리함이 있으나 프라이머 의 길이가 상대적으로 짧아서 비 특이적인 결합을 너무 쉽 게 하기 때문에 재현성이 떨어지는 단점이 있다.
AFLP
AFLP는 RFLP 와 RAPD의 단점을 보완하기 위하여 서 로의 장점을 이용하여 재현성이 높으면서 PCR 기반으로 개발 된 분자마커이다(Farooq & Azam, 2002). RFLP처럼 제한효소로 잘라준 작은 크기의 DNA를 염기서열을 알고 있는 어댑터(adaptor)를 붙여준 뒤, 선택적 PCR(selcetive PCR)을 이용하여 DNA 조각을 증폭하여 다형성을 검정하 는 방법이다(Vos et al., 1995). 각 프라이머 세트별로 대개 1번의 AFLP 분석에서 50~100 개의 밴드를 생성한다. AFLP 의 가장 큰 장점은 많은 수의 DNA 다형성을 만들어 낼 수 있으며, 분석하려고 하는 작물의 DNA 염기서열에 대 한 정보가 요구되지 않고, PCR 기반 분자마커이기 때문에 매우 빠르게 진행할 수 있다는 것을 들 수 있다(Farooq & Azam, 2002). 대부분의 AFLP 마커는 유전체상에서 고유의 위치를 가지기 때문에 물리적 지도(physical mapping)에 이 용될 수 있으며, 이를 활용하여 서로 가까운 종이나 아종 간 의 근연 관계를 분석 할 수 있다(Yin et al., 1999; Rouf Mian et al., 2002).
염기서열 특이적 PCR 기반 마커
Microsatellite 기반 마커
VNTR(Variable Number Tandem Repeat)는 짧은 염기 서열 구간이 직렬로 반복되어 있는 구간을 말하며 VNTR은 많은 염색체에서 발견되는 각 개인마다 반복되는 길이의 차 이가 큰 부분이다. 기본적으로 VNTRs는 microsatellites와 minisatellites로 구분지어 지는데 일반적으로 (TG)n 또는 (AAT)n 같이 2개~10개 사이의 염기서열이 반복되는 것을 microsatellite [또는 SSR(Simple Sequence Repeat), STR (Short Tandem Repeat)와도 통용하여 쓰기도 한다.] 이라고 부르며 minisatellite는 기본적으로 반복되는 단위가 더 길 때 사용된다. 요즘은 microsatellite는 반복되는 구간이 5 bp 이하를 의미하고, minisatellite는 10~60 bp 정도의 반복 구 간을 의미한다. 개체의 확인과 집단 내 또는 집단 간의 유전 적 변이를 연구하기 위해 이상적인 마커는 매우 높은 변이 를 보이는 하나의 유전자좌이다. Minisatellite 염기서열을 제공하는 유전자좌에 대한 특이적 probe의 개발은 이러한 매우 다양한 VNTR에 대한 대립유전자적 분석을 가능하게 해준다. 그러나 대부분의 집단 및 생물학적 다양성 연구에 서는 일반적으로 minisatellite보다 microsatellite이 이용된 다. Microsatellite 나 STR, SSR은 진핵생물이 가지고 있는 염기서열의 반복이 DNA 복제 과정 중 반복 구간의 절단이 나 삽입에 의해 변이가 생기게 되는데, 바로 이 차이를 이용 하여 분석하는 방법이다(Schlötterer & Tautz, 1992). Microsatellite DNA는 한 유전자좌에 약 30~50개의 대립 유전자를 조사할 수 있기 때문에 집단 유전 연구에 흔히 쓰 이고 있다. Microsatellite 마커 개발에 있어서 초기의 많은 어려움에도 불구하고 많이 이용되는 것은 독립적인 유전자 좌에 대한 대립형질의 변이에 대해 연구할 때 매우 큰 이점 이 있기 때문이다. 그러나 microsatellite의 가장 큰 단점은 프라이머를 개발하는데 많은 시간과 노력이 들어간다는 사 실이다(Parker et al., 1998; Mueller & Wolfenbarger, 1999). 이러한 단점으로 인해 하나의 종에서 개발된 microsatellite 마커는 매우 근접한 종 이외의 종에서는 거의 사용 할 수 없는 경우가 자주 발생하게 된다. 그러나 일반적으로 새로운 종에서 microsatellite를 이용하여 연구를 할 경우 특 이적 프라이머를 새로 개발하는 것이 가장 유리하나 경우에 따라 이미 개발되어 사용 가능한 가장 가까이 연관된 종의 프라이머를 사용할 수 있다.
ISSR(Inter-simple Sequence Repeat)
ISSR 마커는 서로 반대 방향의 동일한 두개의 microsatellite 사이를 PCR 증폭하여 그 차이를 분석하는 기술이 다. 이 방법은 다양한 유전자 좌를 목표로 하는 단일 프라이 머를 이용한 PCR 반응으로서 microsatellite을 이용하여 서 로 다른 길이의 inter-simple sequence repeats를 증폭시키는 기술이다. PCR 산물들은 역위(inverted)의 microsatellites에 의하여 거의 무제한적이며, 일반적으로 높은 다형성을 보이 기 때문에 다양한 변이 분석에 용이하게 이용될 수 있다. 또 한 ISSR은 RAPD처럼 매우 간단하나 RAPD에서 사용하는 프라이머(10 mers) 보다 긴 프라이머(15~30 mers)를 이용 하기 때문에 좀 더 높은 온도에서 정교하게 사용할 수 있다. ISSR 마커는 microsatellite 마커의 특성을 가지고 있으면서 프라이머 합성 시에 염기서열 정보가 필요하지 않기 때문에 손쉽게 합성 할 수 있다는 장점이 있다. ISSR은 간단하고 빠르며 방사선 동위 원소가 필수적이지 않기 때문에 다소 안전한 편에 속한다. 그러나 RAPD처럼 재현성과 우성 유 전이 ISSR의 가장 큰 한계점이다.
SNP(Single nucleotide polymorphism)
SNP는 집단내의 각 개체 간의 유전체의 염기서열에 존재 하는 한 염기쌍의 차이(single base-pair variation)에서 발생 하는 다형성(polymorphism)을 말한다. 그렇기 때문에 가장 많이 존재하는 분자 마커이며, 비록 각 종간에 발생과 분포 에 변이가 있기는 하나 SNP는 유전체 전체에 퍼져있는 분 자 마커이다. 일반적으로 SNP는 non-coding 부위에서 더 많이 발견되며, 보통 1 kb 당 1개의 SNP가 존재한다고 알 려져 있으나 옥수수의 경우에는 60~120 bp 마다 1개의 SNP가 존재한다고 보고되어 있다(Ching et al., 2002). 이처 럼 식물체 유전체에서는 빈번한 SNP 다형성 때문에 mapping, MAS 그리고 유전자 지도 기반 클로닝(map-based cloning) 이 가능하게 되었다(Batley et al., 2003). 염기 서열 분석 기 술의 발달과 EST(Expressed Sequence Tag) 데이터베이스 의 축적으로 SNP 마커 개발이 더 활발해졌다. 그로인해 SNP 는 기존 마커들에 비해 고밀도의 유전자 지도를 작성 할 수 있게 되었다. 또한 DNA chip, 대립유전자 특이 PCR (allele specific PCR), 프라이머 확장(primer extension) 등을 이용한 high-throughput 기술을 이용하여 마커 개발의 전 과 정이 고속으로 자동화 될 수 있다는 점이 다른 분자 표지들 과 차별화된 특성이라 할 수 있다(Agarwal et al., 2008).
응용 분자마커(Advanced Molecular Marker)
SCAR(Sequence Characterized Amplified Regions)
SCAR 마커는 짧은 시간에 목표로 하는 유전자에 연관된 마커를 이용하여 선발과 유전자 지도 기반 클로닝에 이용하 기 위하여 개발되었다. SCAR는 한 쌍의 특이적인 프라이 머를 이용하여 PCR로 증폭된 이미 유전적으로 정의된 유 전자 좌의 DNA 절편을 말한다. 이 절편은 클로닝을 통하여 염기서열 분석 후 DNA 절편 양끝에서 특이적으로 제작된 15~30 bp의 프라이머를 이용하여 SCAR 마커로 개발할 수 있다(Paran & Michelmore, 1993). 이때 개발된 SCAR 마커는 PCR 기법에서 처음 클로닝했던 것과 크기와 비슷 한 단일 PCR 밴드가 나와야 한다. SCAR 마커는 STS (Sequence-Tagged Site) 마커와 매우 유사하다. STS 마커는 200~500 bp 의 짧은 단일-copy의 DNA 절편으로서 한 쌍 의 프라이머를 이용한 PCR 에 의해 고유의 결과물을 증폭 해 낼 수 있는 마커를 말한다. 이런 고유의 특성 때문에 STS는 방사선 동위원소 hybrid mapping과 연관에 자주 이 용되며 유전체의 물리지도(physical map)에서 DNA 표시 인자 역할을 하는데 STS는 기본적으로 중복되는 부분을 가 지고 있지 않는다(Olson et al., 1989). 반면에 SCAR은 STS 의 장점을 공유하면서도 2가지 측면에서 차이가 있는데, 첫 째는 SCAR는 우선적으로 유전적으로 정의되어 있기 때문 에(primarily defined genetically) 유전체에서 물리적 표시 인자(physical landmarker) 뿐만 아니라 유전적 마커로서의 기능도 할 수 있으며, 둘째는 SCAR은 오직 PCR에 의해서 만 분석되기 때문에 반복되는 구간을 가지고 있다는 특징이 있다(Paran & Michelmore, 1993).
CAPS(Cleaved Amplified Polymorphic Sequences)
CAPS 마커는 RFLP 마커를 PCR 마커로 전환하여 시간 과 노동력을 절감할 수 있는 마커로서 PCR-RFLP 마커라고 도 불린다. CAPS 마커는 SNP처럼 한 개의 염기서열이 변 하거나 InDel에 의해 발생하는 제한효소에 의해 잘리는 부 위의 변화를 해석할 수 있는 마커이다(Chelkowski & Stephen, 2001). CAPS 마커는 유전자좌에 특이적인 프라이머로 PCR 증폭을 한 후 제한효소로 잘라준 뒤 나타나는 다형성 을 분석하는 방법이다. 그렇기 때문에 SSCP, SCAR, AFLP, RAPD 등과 연계하여 다양하게 활용할 수 있다. CAPS 마 커는 공우성 마커이고 대립유전자 특이적 마커이기 때문에 식물에서 해당 대립유전자의 동형접합자와 이영접합자의 관계를 확인할 수 있다. 그렇기 때문에 CAPS 마커는 genotyping, positional cloning이나 유전자 지도 기반 클로닝(map-based cloning)에 유용하게 이용된다(Spaniolas et al., 2006).
RAMP(Randomly Amplified Microsatellite Polymorphisms)
Microsatellite 기반 마커들은 높은 대립형질 특이적 다형 성을 얻을 수 있으나 매우 노동 집약적인 마커이다. 반면 RAPD 마커는 저렴하고 쉽게 개발 할 수 있으나 낮은 다형 성을 보이는 마커이다. 이 두 가지를 보완하기 위하여 나온 것이 Wu et al.(1994)에 의해 개발된 RAMP 마커로서 단순 하게 반복되는 염기서열의 바로 옆의 염기서열의 임의의 분 포를 기반으로 개발되었다. RAMP 마커는 5´ 방향의 anchor 프라이머와 3´ 방향의 반복 프라이머로 구성되어 있다. 반 복되는 프라이머 방향은 labeling 되어 있기 때문에 anchor 프라이머에 의해 증폭된 PCR 결과물 만 확인 된다.
SRAP(Sequence-Related Amplified polymorphisms)
SRAP 마커의 목표는 ORF(Open Reading Frame)을 증폭 하는 것이다. 기본적으로 17~21 bp 의 길이를 가지는 임의 의 2개의 프라이머를 이용하여 PCR 증폭 시키며, AT나 GC의 함량이 높은 부분을 증폭시키도록 구성된다(Li & Quiros, 2001). SRAP 마커를 위한 프라이머 제작에는 2가 지 중요한 요인이 있다. 첫째는 핵심 염기서열이라고 부르 는 13~14 bp 의 길이로서 그 중 처음 5´ 말단 부분의 10~ 11 bp은 특별히 중요한 구성 요소가 없으며 이 부분을 filler 라고 부른다. 둘째, 핵심 염기서열 이후에는 정 방향 프라이 머에는 CCGG가 있으며, 역방향 프라이머에는 AATT 로 구성되어 있다. 핵심 염기서열 뒤에는 3´ 말단에 3개의 선 택적 염기서열(selective nucleotide)이 존재 한다. 그리고 한 쌍의 프라이머에서 filler 부분의 염기서열은 반드시 달라야 한다. PCR 프로그램에서는 처음 5 cycle 에서는 annealing 온도가 35°C이고, 이후 35 cycle 에서는 50°C에서 수행한 다. SRAP 마커는 단순성, 재현성, 적정 수준의 대량 분석 정도와 염기분석의 편리성의 조합된 마커이다(Li & Quiros, 2001). SRAP 마커는 ORF를 목표로 하기 때문에 염기서열 의 삽입과 결실을 이용하여 적절한 수의 공우성 마커 개발 이 가능하다. SRAP 마커 시스템은 여러 가지 목적으로 서 로 다른 작물에 적용되어 왔다(Gulsen et al., 2007).
TRAP(Target Region Amplified Polymorphisms)
TRAP 마커는 생물정보학과 EST 데이터베이스를 이용한 빠르고 효율적인 PCR 기반 마커이다. TRAP 마커는 18 bp 짜리 2개의 프라이머를 이용하여 한 개의 프라이머는 목표 로 하는 유전자의 EST 데이터베이스를 기반으로 구성되는 고정 프라이머(fixed primer)이며, 다른 하나는 AT 나 GC 비율이 높은 부분에서 인트론이나 엑손과 결합하도록 구성 된 임의의 프라이머를 이용한다(Hu & Vick, 2003). TRAP 마커는 특정 유전자 염기서열에 대해 개발된 마커이기 때문 에 germplasm 간의 genotyping이나 농업적으로 유용한 유 전 형질에 대한 마커로서 육종에 활용하는 데 많이 이용되 고 있다(Hu et al., 2005).
SSCP(Single strand conformation polymorphism)
SSCP는 한 개의 염기서열 변화와 같이 염기서열의 미묘 한 차이 때문에 DNA의 단일 가닥이 서로 다르게 접혀지는 원리를 이용하여 단일 DNA 가닥의 이동성의 변화를 분석 하여 다형성을 확인 하는 마커이다(Orita et al., 1989). 상보 적인 가닥이 없을 때에는 단일 가닥의 내부에서 염기 결합 이 생기지 않기 때문에 고리모양이나 접힘이 생기게 되고 이로 인해 독특한 3차원 구조가 생겨 전기영동을 해보면 젤 상에서 서로 다른 이동성을 보인다. 가끔 SSCP 마커는 같 은 PCR 결과물의 두 DNA 가닥을 분리하여 다형성을 분석 하거나 두 부모 개체에서 동일한 부분의 PCR에서의 다형 성을 분석하는데 유용하게 이용되기도 한다. PCR 기반 SSCP 마커는 빠르고, 간편하며, SNP나 InDel 에 대한 감도 가 높은 마커이다. 그렇기 때문에 점 돌연변이(point mutation) 을 분석해 내는데 유용하게 이용 된다(Fukuoka et al., 1994). SSCP 마커는 대립유전자의 변이를 찾는다는 기술적 인 측면에서 RFLP와 유사하나 DNA 조각의 여러 부분에서 돌연변이와 다형성을 찾을 수 있다는 차이가 있다.
RNA 기반 분자마커
RNA 기반 분자 마커는 기존의 마커의 개념보다는 유전 적 발현에 의한 생물학적인 반응을 의미하는 바가 더 크다. 그래서 RNA 기반 마커를 통한 깊이 있는 유전 정보는 유 전자의 발현에 대한 특이적 발현 양상에 대한 연구를 필요 로 한다.
cDNA-AFLP
cDNA-AFLP는 새로운 RNA 지문법의 하나로 특이적으 로 발현하는 유전자를 발굴하기 위해 사용되는 마커이다 (Bachem et al., 1998). 기본적인 기술은 AFLP 와 유사하나 우선 cDNA를 만들어 준 뒤, 서로 다른 두 개의 제한 효소 로 잘라준 뒤 어댑터를 붙여주고 선택적 PCR(selective PCR)을 통하여 다형성을 분석한다. 분석 집단이나 개체에 서 나타나는 다형성은 각 밴드마다 특이적인 유전자의 발현 을 의미한다.
cDNA-SSCP
cDNA-SSCP는 RT-PCR의 SSCP 분석을 통하여 유전자 의 발현 정도를 평가하는 방법이다. 이 방법을 통하여 배수 성 유전체를 갖는 식물로부터 매우 높은 유사성을 갖는 상 동 유전자에 대한 발현 양상을 검정하는데 매우 유용하게 이용된다. Cronn & Adams(2003)의 보고에 의하면 cDNASSCP를 이용한 유전자 발현에 대한 반복 실험에서 99% 이 상의 동일함이 증명되었다.
RAP-PCR(RNA fingerprinting by arbitrarily primed PCR)
임의의 프라이머(arbitrary primer)를 이용하여 이중 나선 의 cDNA를 합성하고, 합성된 cDNA에서 PCR을 수행하는 방법이다. 이 방법을 통하여 서로 다른 개체의 같은 조직에 서 추출한 RNA 또는 같은 개체의 서로 다른 조직에서 추 출한 RNA에 대한 fingerprinting이 가능해 진다. 이러한 개 체별 차이는 바로 염기서열의 차이이기 때문에 genetic mapping에 유용하게 이용되며, 조직 간의 차이는 유전자의 발현 양상의 차이를 검정하는 것이 이용된다.
Transposable element-based molecular markers
전이인자(transposon)는 유동성을 갖는 유전 요소로서 유 전체 안에서 자신의 위치를 바꿀 수 있다. Transposon 은 60여 년 전에 옥수수에서 처음 발견되었으며 크게 2개의 클 래스로 나눠진다. 클래스 I 전이인자는 우선 DNA에서 RNA로 전사가 일어난 뒤, RNA가 DNA로 역전사가 일어 나게 된다. 이때 복제된 DNA는 유전체의 새로운 위치에 삽 입되게 된다. 이때 역전사 과정에서 사용되는 역전사효소는 종종 전이인자 자체에서 스스로 코딩되기도 한다.
반면에, 클래스 II는 RNA로 전사되는 단계를 거치지 않 고 자신의 위치에서 잘려 나와서 새로운 위치에 들어가는 (cut and paste) 형태를 가지고 있다(Grzebelus, 2006).
역전이인자(Retrotransposon) 기반 분자마커
커다란 유전체를 가진 식물에서는 역전이인자(retrotransposon) 가 전체 유전체의 40~60%를 구성하는 반복되는 DNA의 주요 클래스에 해당 된다(Kumar & Bennetzen, 1999). 역전 이인자는 LTRs(Long Terminal Direct Repeats), LINEs (Long Interspersed Elements), SINEs(Short Interspersed Elements)로 3가지 부분으로 분류되는데 LTR 역전이인자 는 100 bp~5 kb 에 이르는 직접적으로 반복되는 LTR을 가 지고 있으며, LTR 역전이인자는 다시 copia-like, gypsylike, 그리고 BEL-Pao-like groups 으로 유전자의 유사성과 유전자가 코딩되는 순서에 따라 3종류로 분리 된다. 일반적 으로 LTR 역전이인자는 레트로바이러스(retrovirus)와 유전 자의 코딩 단백질이 유사하며 copia-like나 gypsy-like 역전 이인자는 식물에서 많이 발견된다. 역전이인자는 염기서열 의 보전(conserved), 충분한 길이, 그리고 새로운 삽입으로 인한 다형성의 생성으로 분자 마커로 개발하기 유리하다 (Kalendar et al., 1999). Non-LTR 역전이인자에는 LINEs 과 SINEs 이 있다. 이들 역시 식물체에서 25 만개가 넘는 높은 copy 수를 가지고 있으며 진핵세포에서 넓게 분포되 어 있다. LINEs 은 2개의 ORF를 가지고 있으며, 이것들은 역전이 과정에서 필요로 역전사효소와 엔도뉴클리아제 (endonuclease) 기능을 가지고 있으면, 또한 리보뉴크레오단 백질(ribonucleoprotein)을 형성하는데 필요한 핵산의 결합 하는 기능도 가지고 있다. 반면, SINEs 은 역전사효소의 기 능이 없으며 전이를 위한 다른 요인을 따르게 된다.
역전이인자 기반 분자 마커는 한 개의 프라이머는 역전이 인자에 결합하고, 또 다른 하나는 이웃하는 유전체에 결합 을 한다. SSAP(Sequence-Specific Amplified Polymorphisms) 의 경우는 역전이인자가 삽입되어 있는 부분과 어댑터가 결 합되어 있는 부분의 제한효소 부분에 결합하여 DNA를 증 폭시킨다(Waugh et al., 1997). IRAP(Inter-Retrotransposon Amplified Polymorphism)에서는 인접하고 있는 2개의 역전 이인자는 각 LTR 사이의 DNA를 증폭하여 다형성을 분석 하는 기술이다. REMAP(Retrotransposon- Microsatellite Amplified Polymorphism)은 역전이인자의 삽입된 부분과 microsatellite 의 부분을 증폭한다. RBIP(Retrotransposon- Based Amplified Polymorphism)은 역전이인자가 비었거나 발생한 곳을 추적할 수 있다.
TD(Transposable Display)
TD는 AFLP를 변형시킨 방법으로서 많은 copy 수의 계 통에서부터 많은 TE(Transposon Element)를 동시에 확인 할 수 있는 기술이다. 이 기술은 서로 다른 2개의 제한효소 를 이용하나 1개는 genomic DNA 중 일부를 잘라주고, 다 른 하나는 TE 의 회문부분을 잘라주는 제한효소를 이용한 다. 그 뒤 AFLP처럼 어뎁터를 결합 시킨뒤, TIR(Terminal Inverted Repeat)에 결합하는 프라이머나 삽입된 부분에 결 합하는 프라이머를 이용하여 표현형과 부합하는 개체를 선 발하는 마커로 활용한다(van den Broeck et al., 1998).
IMP(Inter-MITE polymorphism)
TD 기술을 활용하여 MITE(Miniature Inverted-repeat Transposable Elements)라고 불리는 특이한 특성을 갖는 TE가 개발되었다(Casa et al., 2000). 이 기술은 원칙적으로 IRAP 와 매우 유사하다. 그러나 IMP는 역전이인자가 아닌 MITE 전이인자를 이용하여 PCR 증폭을 한다. MITE 는 짧 고, 클래스 II 전이인자이기 때문에 저절로 전이 되지 않는 다. 또한 기식물의 유전체에 많이 퍼져있고, 풍부하며 다양 한 크기와 염기를 가지고 있는 많은 copy 가 존재한다.
DArT(Diversity Arrays Technology)
DArT 기술은 genotyping이나 다른 유전 분석을 위한 염 기 서열 수준의 마커를 개발하기 위한 기술이다. DArT는 어떤 개체나 집단의 전체 genomic DNA의 representation에 서 특정 DNA 절편의 존재 여부를 추적하는 기술로서 벼에 서 처음 개발된 기술이다(Jaccoud et al., 2001). 최근에는 애기장대, 카사바, 보리, 수수, 귀리, 밀, 호밀 등 다양한 작 물에서 활용이 되고 있으며, DArT는 개발도상국이나 농업 후진국에서 작물에 대한 유전적 다양성과 유전자 지도를 연 구하도록 많이 장려되고 있다. DArT는 microarray를 기반 으로 하기 때문에 유전체 전체에 분포되어 있는 많은 다형 성 유전자좌를 동시에 분류하는 것을 가능하게 해 준다 (Jaccoud et al., 2001). DArT는 배수체 식물에서 SNP 추적 을 기반으로 하는 다른 array 와 비교하여 확실히 편리한 장 점이 있다(Wenzl et al., 2004). 그 외에 DArT는 연구할 품 종의 참고 염기서열 정보가 필요하지 않다는 장점 이외에도 효율이 매우 좋고 빠르며 재현성이 매우 높으며 SSR 마커 와 같은 다른 분자 마커와 비교해보면 얻어지는 데이터 대 비 소요 비용이 매우 저렴하다는 장점이 있다.
NGS(Next-Generation Sequencing)
그동안의 염기서열 분석은 Sanger 방법(Sanger et al., 1977)을 기본으로 시행되었지만 최근에는 NGS 방법이 제 시되어 많이 활용되고 있다. NGS는 ‘sequencing by synthesis’ 원리를 이용하여 하나의 염기에 대한 서열분석과 합성이 일 어난 뒤 다음 염기의 분석이 순차적으로 진행되는 방법이 다. NGS 방법은 DNA 염기를 증폭 시킨 뒤 형광 표지 인자 를 인식하여 영상화 과정을 통하여 각 DNA의 염기 서열 정보를 분석한다. 그렇기 때문에 분석 장비에 따라 분석할 수 있는 길이(read length), 데이터의 크기와 형식, 그리고 염기서열 분석 방법 등이 모두 다르지만 분석이 병렬식이라 동시에 수백만 개를 분석해 낼 수 있다는 장점이 있다. NGS 기법이 발달하면서 식물 유전자원의 분석을 그동안에 유용하게 활용되던 몇 가지의 분자 마커에 의존하지 않고 식물의 유전체 전체를 읽어주는 분석 정보를 활용할 수 있 게 되었다. 또한 식물의 유전체 분석 정보는 품종 및 유전자 원의 순계 유지에도 활용이 되고 있으며 SNP 마커 개발에 유용하게 이용되고 있다(Davey et al., 2011). 이와 더불어 유용한 유전자원의 다형성 발굴과 양적 형질과 관련된 후보 유전자 군도 발굴해 낼 수 있다(Mackay et al., 2009).
분자 표지 인자와 mapping population을 활용한 옥수수 육종
유전체학의 발달은 지난 십 수 년간 옥수수에서 수많은 DNA 표지 인자를 개발해냈다. 대부분 microsatellite 나 SSR 마커 등이 이용되었으며, 최근에는 SNP나 InDel 마커 등이 활용되고 있다. 더군다나 SSR 마커나 SNP 마커는 식 물발달, 생물/비생물(biotic/abiotic) 스트레스에 저항성, 작 물의 질 등과 같은 부분을 조절하는 수많은 유전자들이 옥 수수에서 발굴 및 발현 검정이 이루어져서 분자 마커를 이 용한 육종에 이용되고 있다(Prasanna et al., 2010). 요즘은 수많은 정보의 공유성과 편리성, 효율성 때문에 SSR 마커 가 옥수수에서 가장 일반적으로 이용되고 있는 표지인자이 다. PCR을 기반으로 하는 유전적으로 공우성 마커들은 강 력하며, 재현성이 좋고, 과변이성이며, 식물체의 유전체에 균일하게 퍼져있다(Powell et al., 1996). SSR 과 SNP 모두 대량으로 분자 육종에 이용되고 있지만 최근에는 SNP가 SSR 보다 자동화 시스템에 적합하기 때문에 유전과 육종의 목표에 더 부합하고 있다. 다른 작물의 유전체와 비교하 여 옥수수의 SNP의 빈도는 매우 높으며, 대략 28~124 bp 의 염기서열 당 1개의 SNP가 발견되고 있다(Tenaillon et al., 2001; Ching et al., 2002; Vroh Bi et al., 2006). 옥수수 에 대한 SNP의 데이터베이스와 자료들은 옥수수의 유전자 형, 표현형, 다형성 자료를 옥수수의 inbred line과 population 에 대하여 정립되어 있다(hao et al., 2006; http://www. panzea.org). 최근에는 여러 개의 대량 유전자형 분석 플랫 폼이 개발되어 백만 개의 SNP 마커를 동시에 분석할 수 있 게 되었으며, 최근에는 1536 SNP가 포함된 맞춤형 GoldenGate assay가 개발되어 옥수수의 SNP 정보의 대중화에 크게 기 여하였다(Yan et al., 2009).
마커 시스템 이외에도 옥수수에서는 B73 X Mo17 의 중 간 종(intermated) 집단(IBM)이나 IBM에서 개발한 RIL (Recombinant Inbred Line)에 대한 다양한 mapping population 을 이용하여 육종에 활용하고 있다(Lee et al., 2002; Coe et al., 2002; Cone et al., 2002). 또 다른 최근 개발된 옥수수 주요 유전자원으로는 NAM(Nested Association Mapping) 집단이 있는데, NAM 집단(population)은 245 개의 집단에 서 200 개씩 총 5000개의 RIL 계통을 구축해 놓은 옥수수 유전자원 집단이다(Fig. 1(A)). NAM집단은 QTL mapping 의 통계적 처리와 거의 유전자 수준에 다다르는 높은 염색 체 수준의 mapping 해상도를 가지는 association mapping 을 조합하여 근원적인 다중 형질에 대한 연구 및 육종에 활 용할 수 있는 옥수수 집단이다. NAM population은 양친 중 1개의 부모계통을 공유하나 각기 다른 두 번째 부모계통을 갖는 세트로 되어있는(nested) 계통이다. 즉, 모든 25개의 family들은 미국 옥수수 육종에서 가장 중요한 B73 계통을 공유하고 있다. B73 계통은 미국 옥수수 분자 육종에서도 중요한 유전자원이기 때문에 옥수수 육종에서 B73의 자식 계통들이 광범위하게 차지하고 있으며, B73의 유전체는 이 미 염기서열 분석이 끝났기 때문에 분자육종 연구에서 다양 하게 활용되고 있는 계통이다(Schnable et al., 2009). NAM 집단의 교배친들은 미국 옥수수 육종에서 농업적으로 중요 하다고 평가받는 inbred line이며, 전 세계에서 수집된 94개 의 SSR 마커로 분석하여 유전적 다양성을 획득하기 위한 계통을 중심으로 선정되었다(Liu et al., 2003; Flint- Garcia et al., 2005; Liu et al., 2008). NAM 집단에 사용된 대표적 인 교배친들로는 Suwan(태국계통), Ki3(한발 감수성), Ki11 (한발 저항성) 등이 교배친으로 이용되었으며, 또한 CIMMYT 에서 개발한 열대성 품종 8개(CML52, CML69, CML103, CML228, CML247, CML277, CML322, CML333) 등이 이 용되었다(Yu et al., 2008). NAM 집단의 유전적 다양성을 고려하여 각 유전자 연관 연결지도(association mapping)가 활용이 되었으나, 옥수수 육종에 RIL을 이용하기에는 가끔 너무 많은 표현형을 보이거나 또는 특정 재배환경에 적응력 이 떨어지는 경우가 있다. 그래서 더욱 지역 특이적 유전자 연관 지도(association mapping)를 실행하여 재배 지역에 적 당한 유전자원을 개발하는 것이 중요하다. 최근에는 중국에 서 유전자 연관 지도(association mapping)를 이용하여 온대 지역 적합 germplasm을 갖는 155 inbred 계통을 개발하는 등 분자 마커가 육종에 다양하게 이용되고 있다(Yang et al., 2010).
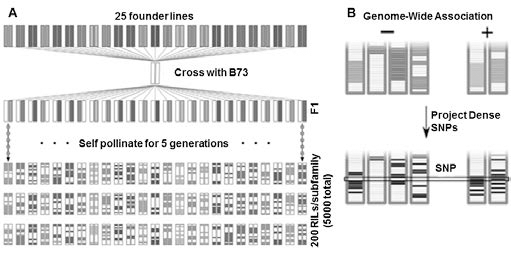
Fig. 1.
Schematic design of Nested Association Mapping (NAM). (A) The maize NAM population was created by crossing 25 founder lines by the B73. Single-seed descent and self-pollination for five generations were then used to generate 200 recombinant inbred lines (RILs) for each subfamily. (B) Genome-Wide Association uses the information of which chromosomal segments were inherited from which parent (top) to project dense genotyping from the founder lines onto the progeny for improved resolution (bottom). Colored bars show which parent the chromosomal segment originated from. Single-nucleotide polymorphisms (SNPs) in panel c are shown as either matching the allele present in B73 (white) or as the alternative allele (black). Figure based on Wallace et al. (2014).QTL mapping
방대하면서 쉽게 눈에 보이지 않는 DNA 수준의 가변성 의 발견은 분자마커 분석과 양적형질에서 일치를 보이는 변 이 분석에 대한 통계 패키지의 발달과 더불어 옥수수 및 다 양한 작물에서 농업적으로 중요한 형질의 배열에 영향을 미 치는 QTL mapping으로 발전되어왔다. QTL은 양적형질에 연관(association)되어 영향을 끼치는 유전체의 지역으로 정 의되는데 개념적으로 QTL은 한 개의 유전자나 혹은 형질 에 영향을 끼치는 단단히 연관(likage)된 군집이다. QTL mapping과 분자 마커 발굴은 목적형질에 주도적으로 영향 을 끼치는 QTL과 매우 가깝게 연관되어 있어서 여교잡, 계 통도, 집단의 유전적 향상을 위한 MAS에 이용된다. 1987 년에 수량 관련 형질로 옥수수 QTL이 실시 된 후 전 세계 의 수많은 옥수수 연구자들이 농업적으로, 과학적으로 필요 한 다양한 형질에 대한 유전자/QTL에 tagging 되는 분자 마커를 개발하기 시작하였다(Stuber et al., 1987). 분자 마 커는 수량, 순화, 환경적응, 병해충 저항성, 한발 및 열 스트 레스 저항성 등 다양한 형질에 연관된 QTL을 발굴하여 특 성 검정을 위하여 사용되어 왔다. 이러한 정보는 Maize DB(http://agron.missouri.edu)를 이용하여 확인해 볼 수 있 다. 여기에서는 옥수수 재배를 제약하는 두 가지 가장 중요 한 생물학적 스트레스와 환경적 스트레스인 downy mildew 와 한발 스트레스에 대한 QTL 분석에 대하여 알아보도록 하겠다.
Drought tolerance 육종
한발 스트레스는 옥수수 생산과 육종에서 가장 큰 제약을 주는 스트레스이다. 그렇기 때문에 특히 남동아시아 지역에 서는 한발저항성 연구가 최우선되고 있다. 옥수수는 특히 영양 생장기 동안 물 부족으로 인한 스트레스에 매우 민감 한데, 그 이유는 이 시기의 한발 스트레스가 바로 수량과 직 결되기 때문이다. 분자마커 시대의 도래로 인하여 스트레스 와 비 스트레스에 조절되는 목적 형질에 관여하는 유전체 부분의 발굴이 가능해 졌다. QTL은 스트레스 조건 하에서 는 한발 저항성과 관련된 유전체의 집단을 찾을 수 있게 하 였다. 한발 저항성 관련 유전자들도 수분 공급이 제대로 될 때에는 식물 생육에 관련하는 것을 조절하는 것으로 알려졌 다. 그럼에도 QTL 집단들은 수분 공급이 제대로 될 때와 안 될 때 모두 발견되는데 그 이유는 식물이 수분 환경에 대해 광범위하게 적응하도록 관여하기 때문이라고 유추되 고 있다(Ribault et al., 2008).
옥수수에서 QTL 분석을 이용하여 한발 저항성에 대한 유전분석이 1995년 처음 실시된 이후에(Lebreton et al., 1995), 옥수수에 수분 공급이 제한된 상태에서 수량이나 중 요한 생태적 생리 형질을 조절하는 QTL을 발굴하기 위한 연구가 많이 진행되었다. 또한 여러 변이 품종에서의 QTL 연구를 통하여 단일 유전자 지도를 엮어서 옥수수 유전체에 걸쳐 분포되어 있는 한발 스트레스에 관련이 있는 QTL를 찾아냈다(Tuberosa et al., 2002b). 옥수수와 그 외 다른 작 물들의 비 생물적 환경스트레스 저항성과 연관된 주동 유전 자와 QTL 데이터는 웹사이트를 통하여 확인할 수 있으며 (Plantstress: http://www.plantstress.com/biotech/index.asp? Flag=1), Plantstress에 수집된 데이터를 기반으로 옥수수의 한발 스트레스 관련 형질에 대한 QTL 자료들은 Table 1 에 정리되어 있다.
Table 1.
Summary of QTLs identified for drought tolerance-related traits in maize.
Khavkin & Coe(1997)는 옥수수에서 큰 영향을 미치는 많은 QTL들은 사실 항상성 유전자와 전사 인자 유전자와 같은 유전자의 집단이며, 이 유전자 집단을 이용하여 식물 발달과정이 조절되며, 또한 이런 유전자 집단들에 의해 많 은 식물이 비 생물학적 스트레스에 반응한다고 추측하였다. 옥수수에서 한발 저항성 QTL 연구와 육종 프로그램에서 MAS에 QTL를 적용시키기 위한 추진 전략은 많은 논문과 연구에 진행되었다(Araus et al., 2008; Collins et al., 2008). 한발 스트레스가 쉽게 일어나기 쉬운 환경에서 옥수수 생육 의 유전적 분석은 DNA 마커의 이용을 통하여 많은 유리함 을 얻을 수 있는 반면(Messmer et al., 2009; Szalma et al., 2007), 지금까지 실질적으로 옥수수 육종 프로그램에 직접 적용되는 경우는 별로 없었다. QTL은 각 QTL의 원래의 유 전적 배경과 목적 형질의 단일 유전자의 영향에 특이적이기 때문에, 많은 수의 마커를 활용한 육종 실험에서 한발 저항 성을 증진시키는 것이 제한적이다. 이런 현실적 괴리는 유 전적 바탕의 복잡성과 유전적 배경의 영향 때문이며, 식물 발달 단계와 환경에 QTL이 영향을 받는 것, 부적합한 표현 형의 fine mapping을 진행하는 과정에서의 시간과 비용적 측면, 그리고 유전자와 유전자 사이의 영향 등에 의해 생긴 다(Tuberosa et al., 2002b; Campos et al., 2004; Xu et al., 2009).
최근에는 한발 저항성에 대한 분자 육종의 개념적인 구상 이 제시되고 있다. 이 개념은 수량을 나타내는 Passioura 방 정식에 기반을 둔 것으로 수분의 이용(WU), 수분 이용 효 율(WUE), 수확지수(HI)을 이용하여 수량을 설명하는 것으 로 Salekdeh et al.(2009)에 의해 제시되었다. 이 방정식의 QTL과 Omics 연구에서의 활용은 각 QTL실험에 의해 제 공되는 정보의 균일성을 놀랄 만큼 증진 시킬 수 있을 것이 며, CMTV 같은 소프트웨어나 MetaQTL을 이용한 안정적 인 다중 집단이나 환경에 대한 QTL을 실현 가능하게 할 수 있다(Sawkins et al., 2004). McMullen et al.(2009)은 두 접 근법의 장점들을 조합하여 NAM 집단을 개발하였다. 이 NAM 집단을 이용하여 Buckler et al.(2009)은 옥수수의 개 화 시기의 유전적 변이를 연구하였는데, 개화 시기의 차이 는 많은 작은 QTL들의 효과가 누적된 결과이며, 유전적 상 위성이나 환경적인 효과는 거의 없으며, 어떤 단일 QTL 도 2일 이상의 개화 시기의 차이가 나는 변이를 설명할 수 가 없다고 결론지었다. NAM 집단은 옥수수에서 높은 해상도 의 유전자 지도를 작성할 수 있는 매우 중요한 유전적 도구 임에도 한발 저항성 연구에서 이용하기에는 부모 계통의 염 기 서열 분석과 유전적 다형성 분석, 그리고 5000 개나 되 는 RIL에 대한 한발 저항성에 대한 표현형의 분석이 결코 쉬운 일이 아니라는 문제점이 존재한다. 대부분의 이전의 한발 저항성 QTL 연구는 옥수수에 존재하는 자연상의 변 이체와 목적형질에 대한 표현형의 변이를 최대화하기 위한 육종적 가치가 없는 한 개의 부모 계통을 사용한 많은 변이 체 를 중심으로 연구하였고, 그로 인해 육종 프로그램에 MAS를 이용하는데 발견된 QTL의 이용성에 제약이 있었 다(Ali et al., 2011). 이런 문제점을 극복하기 위하여 엘리 트 육종 계통에서 화학적 돌연변이에 의해 생성된 돌연변이 체에 QTL을 활용한 연구가 진행되었다. 또한 한발 저항성 에 대한 옥수수의 QTL mapping 은 230 개의 CIMMYT에 서 개발된 RIL을 통해서 주동 QTL 이 1, 2, 8, 10번 염색체 라는 것을 알아내게 되었다(Prasanna, 2009). RIL 데이터 분석을 통하여 염색체 1, 2, 8, 10번에 같이 존재하는 한발 스트레스에서 특이적 형질에 영향을 끼치는 QTL 도 찾아 내었다. 또한 다른 주요 QTL 보다 한발 스트레스에서 옥수 수 이삭 당 종자의 개수에 영향을 키치는 확실한 양성 상위 성 QTL 도 발굴되었으며, 중국에서 한발 저항성으로 가장 많이 재배되는 X178의 F2:3 집단에서 ASI(Anthesis-Silking Interval)과 식물개체 당 이삭 숫자에 대한 주동 QTL을 발 굴해 냈다(Xiao et al., 2005; Hao et al., 2008). 이 QTL 들 은 염색체 1번(bin 1.03)과 염색체 9번(bins 9.03∼9.05)에 위치하고 있으며 이들은 한발 스트레스에서 다른 실험에서 발굴된 주동 QTL과 일치한다(Tuberosa et al., 2007). 이와 같이 서로 다른 mapping 집단을 이용한 서로 다른 실험에 서 발굴되는 한발 저항성 옥수수에 대한 동일한 QTL 들로 판명되는 것은 발굴된 QTL 마커가 수분 부족 조건에서 옥 수수 생산량을 늘리기 위한 MAS 에 이용될 가능성이 매우 높은 QTL 후보군이라고 할 수 있다(Prasanna et al., 2010).
Downy Mildew 저항성 육종
전 세계적으로 옥수수의 수량과 생산성에 영향을 끼치는 여러 병원균 중에 노균병(downy mildews)가 병원균의 지리 적 분포와 수량의 감소에 대한 잠재적 능력의 면에서 가장 심각한 병원균으로 생각되고 있다(Nair et al., 2005). 노균 병의 감염으로 인한 가장 큰 옥수수 수량의 손실은 필리핀, 대만, 인도네시아, 태국, 인도, 서아프리카, 베네수엘라, 일 본, 오스트리아, 유럽, 북아메리카 등에서 기록되고 있다 (Bonde, 1982). 노균병은 열대와 아열대 지역에서 두드러지 게 발병하며, 전 세계적으로 열대의 저지대와 아열대, 중간 고도의 점이지대, 고지대에서 옥수수 재배지의 30%에서 노 균병 감염으로 경제적 손실을 격고 있다(Jeffers et al., 2000). 특히 아시아에서는 노균병이 옥수수 생산량을 저해 시키는 가장 중요한 생물학적 스트레스로 여겨지고 있다 (Agrama et al., 1999; Jampatong et al., 2013). 동남아시아 의 가장 문제가 되는 노균병 균주로는 인도의 Peronosclerospora sorgi(수수노균병, sorghum downy mildew)와 P. heteropogoni (라자스탄 노균병, Rajasthan downy mildew), 인도네시아 의 P. maydis(자바노균병, Java downy mildew), 태국의 P. zeae, 필리핀의 P. philippinensis 등 이 있다(Prasanna et al., 2010). 노균병에 저항성을 갖는 Ki3와 감수성인 CML139 를 양친으로 사용하여 개발된 RIL을 이용하여 동남아시아 의 아열대 지역의 5가지 노균병의 저항성에 연관된 QTL이 mapping 되었다(George et al., 2003). 5개의 주요 노균병에 확실히 저항성을 갖는 QTL 연구는 6번 염색체(bin 6.05)의 QTL 이 가장 중요하여 5개의 모든 노균병에 대한 저항성 에 영향을 미치고, 수수노균병과 라자스탄노균병의 저항성 에 대해 20~31% 정도의 표현형의 변이를 보인다. 6번과 3번 염색체에 각각 존재하는 2개의 주동 QTL은 수수노균 병에 감수성인 CM139와 수수노균병 저항성인 NAI116을 양친으로 하여 개발된 여교배 집단을 이용하여 QTL을 검 정하였다(Nair et al., 2005). BC2F1과 BC2F2 의 세대에서 두 개의 주동 QTL에 대한 중요한 선발과 54개 SSR 마커를 이용한 반복된 부모의 유전체에 대한 여교배 선발과 BC2F3 세대에서 인위적으로 감염을 시켜 수수노균병에 대한 표현 형을 선발하는 MAS 방법에 의해 수수노균병에 저항성을 갖는 CM139 계통과 여러 QTL-NIL을 개발해 내었다 (Prasanna, 2009). QTL-NIL 계통과 같은 MAS에서 개발된 계통은 노균병 저항성 옥수수 잡종 개발에 이용되었고, 노 균병 스트레스에서에 대한 전사체 프로파일링 연구가 진행 되었다(Singh et al., 2009). 최근에는 노균병에 저항성을 갖 는 Nei9008(태국품종)과 감수성인 CML289(CIMMYT)의 교배를 통해 나온 251개의 F2:3 계통에 대하여 SSR 마커를 이용하여 QTL 분석을 한 결과 수수노균병의 저항성 관련 하여 9개의 QTL을 발굴 하였다(Jampatong et al., 2013). 각 QTL 들은 2, 3, 4, 6번 염색체상에 각 1개씩 존재하며, 염색체 5번에 3개, 염색체 9번에 2개가 존재한다. Jampatong 등(2013)은 수수노균병에 저항성 QTL 지도상에 참고 문헌 을 통해 다른 병해충 저항성 관련 QTL을 겹쳐서 나타내었 다(Jampatong et al., 2002; Sabry et al., 2006, Phumichai et al., 2012). 그 결과 서로 매우 유사한 위치에 존재한다는 것을 알 수 있는데, 이 결과를 통해 옥수수에서 병해충저항 성 관련 유전자들은 유전체 전반에 걸쳐 자유롭게 분포되어 있는 것이 아니라 군집을 이루어 위치한다는 것을 의미한 다. 노균병 저항성 품종에 대한 연구와 메타락실(metalaxyl) 과 같은 진균제의 살포에도 불구하고, 습한 아열대, 열대 지 대를 중심으로 노균병 피해가 계속되고 있다(Lukman et al., 2013). 종자 처리를 위한 진균제의 처리 비용과 그로 인 한 발생할 수 있는 내성 병원균 문제들이 도래하고 있다. 이 런 문제를 해결 하기 위해서는 좀더 가격 효율적이고, 환경 안정적인 방법을 이용한 저항성 품종을 개발하는 것이 중요 하다.
Association Mapping
앞에서 설명한 것처럼 QTL mapping은 식물이나 동물에 서 복잡한 양적 형질의 유전을 연구하는데 매우 유용하고 잘 정리된 연구 방법이다. 특히 연관지도(association mapping) 은 새로운 유전자나 혹은 관련될 것으로 예측하는 유전자의 검정을 위하여 매우 유용하고 효율적인 방법으로 이용되고 있다(Altshuler et al., 2008; Hunter and Crawford, 2008). 특히 식물에서는 동물보다 더 유용성이 뛰어나기 때문에 작 물 육종 분야에서 활용도가 점차 증가하고 있는 추세이다 (Zhu et al., 2008). 일반적인 유전자 연관지도(linkage mapping) 와는 다르게 association mapping 은 주어진 유전자 집단에 서 높은 해상도로 모든 재조합체나 돌연변이체를 분석할 수 있다(Yu & Buckler, 2006). 하지만 association mapping은 육종에서의 높은 유용성을 가지고 있음에도 불구하고 일반 연관지도(linkage mapping)에 비해 빈도가 낮은 대립유전자 에서는 활용 가치가 낮은 편이다(Visscher, 2008). LD(linkage disequilibrium)은 2개 혹은 그 이상의 유전자 좌에서 대립 유전자들이 비 임의적으로 연합(non-random association) 되어 있는 현상을 말한다. 이런 현상은 같은 염색체에 존재 하는 대립유전자의 집단에 관한 내용으로 정의되어 있는데, 비록 LD가 집단을 기반으로 나타나는 현상(population-based phenomenon)이라 할지라도 일반적으로 더 가깝게 위치한 대립유전자 사이에서는 높은 LD를 갖는 것으로 나타나는 경향이 있다. 그래서 임의의 대립유전자 사이의 연합에서는 연관이 줄어들고 그로인해서 소위 불균형(disequilibrium)이 라고 불리는 현상이 나타나게 된다. 그러나 GWA(Genome-Wide Association)의 기술발달과 그로인한 그 데이터의 증가는 상 업적으로 옥수수 수집 종에서 LD decay(주어진 두 대립유 전자 사이에서 다시 random association 으로 돌아오는 비 율)가 상대적으로 낮다는 것을 알게 되었다. 그러나 두 개의 서로 분화된 옥수수 품종에서 LD decay 는 각 재료들 사이 의 높은 재조합 비율 때문에 1~10 kb 정도의 짧은 간격에 서도 빠르게 일어난다(Gore et al., 2009; Yan et al., 2009). Linkage mapping과 association mapping 연구 모두 표현형 의 변화나 또는 둘 사이의 물리적 거리가 충분히 가깝게 연 관되어 있기 때문에 육종가가 분화된 집단에서 그 대립형질 을 전형적으로 선발하고 다를 수 있는 기능을 갖는 염기 변 이체(대립유전자)를 찾아내는데 활용 할 수 있다. 전통적인 연관지도 연구는 분리되는 각 개체가 수백, 수천 개의 분자 마커를 이용하여 유전자형을 분석하는 것인데, 이 때 이들 마커가 스스로 기능을 갖는 유전자이거나 또는 유전자와 매 우 가깝게 연관될 가능성은 실제로 매우 낮다(Yan et al., 2010). 염색체의 물리적 거리와 더불어 유전적 부동, 자연 선택, 인공선택, 교배시스템, 다른 집단과의 혼합 등으로 인 하여 LD의 변형(breakdown)에 영향을 준다(Flint-Garcia, 2003; Yu & Buckler, 2006). 여러 통계적 매개변수가 LD의 규모를 추정하는데 이용된다고 알려져 있다(Hedrick, 1987). 가장 일반적으로 두 개의 주어진 다형성의 위치에 있는 대립유전자 상태 사이의 상관관계를 예측하기 위하여 r2이 사용된다. 옥수수에 적용된 연구를 분석해 보면 LD decay 의 범위는 원시 품종(landrace)는 1 kb 보다 작게 나 타나는 것부터 엘리트 육종 계통에서는 100 kb 이상이 되 는 넓은 범위를 가지고 있다(Ching et al., 2002). 이러한 상 관관계에서 연관 분석(association analysis)는 해상도가 연 관지도패널(association mapping panel)의 선택에 의해 조절 되어 질 수 있기 때문에 옥수수에서 특히 유용하다. 다시 말 해 더 좋은 엘리트 계통에서는 LD가 높고, 더 많이 분화되 거나 외국에서 들어온 계통들에서는 LD가 낮다는 것을 보 여준다(Yan et al., 2010).
LD는 샘플 사이즈가 이미 사용된 50 개의 개체들 보다 작으면 과평가 되어 있는 것으로 실제 유전적 거리(genomic distance)가 더 커지게 된다. LD decay도 서로 다른 염색체 상 위치에서 넓은 변이를 보인다. 이것은 재조합 율이 염색 체를 따라서 큰 변이가 있다는 것을 보여주는 것으로 염색 체에서 재조합 율이 높고, 동원체에서는 재조합 율이 낮다. 또한 높은 재조합 율은 역전이인자가 삽입되었다는 것을 의 미 하는 것이기도 하다(Dooner & He, 2008). 간혹 매우 광 범위한 LD가 강력하게 선택적 분석된 지역에서 발견되기 도 한다(Tian et al., 2009). 옥수수의 종실에서 카르테노이 드 생산을 조절하는 Y1 유전자 주변에서 그런 예가 발견되 기도 하는데, 이 유전자 주변의 높은 수준의 염기서열의 다 양성이 흰색 옥수수 품종에서 발견되었으나 현대의 노란색 품종에서는 발견되지 않는다(Palaisa et al., 2003; Fu et al., 2010). 이런 표현형적 형질을 이용하여 인간이나 동물에게 카르테노이드의 건강상의 장점으로 인하여 주도적으로 선 발되어지고 있으며 이런 선발이 가능하게 된 이유는 노란색 옥수수의 Y1 유전자 주변 LD가 수백 kb 정도의 거리들을 두기 때문이다. 또 다른 10번 염색체의 일부 부분에서는 1 Mb가 넘는 지역을 조절 할 수 있는 긴 LD를 포함하고 있 는데 이것은 최근 재조합이 결여되거나 선발 때문에 염기서 열의 다양성이 결여되었다는 것을 의미한다. 그러나 근원의 유전자는 아직 밝혀지지 않고 있다(Tian et al., 2009). 27 개의 다양한 inbred line의 서로 다른 길이의(수천~수백만 bp)의 100개 이상의 LD 블록이 옥수수 유전체에서 발굴되 었다(Gore et al., 2009). 어떤 세트의 germplasm에 대한 서 로 다른 길이의 LD 블록들 안에서 목표 유전자의 기능상 돌연변이에 매우 가깝게 연관되어 있는 분자마커를 발굴해 내는 것은 아마 불가능할 것이다. 이런 이유 때문에 목표 유 전자 또는 그 주변의 재조합이나 돌연변이 수를 최대화하기 위하여 적절한 germplasm의 선택은 assocaition analysis 의 성공을 위한 매우 중요한 요인 중의 하나이다(Yan et al., 2010). 일반적으로 유전적 연관지도(linkage map) 연구는 교배가 되고, 오직 몇 세대의 재조합만 일어나서 양친의 mapping population 을 만들었을 때, 분자 마커보다 약간 더 떨어져 있는 실제 발현되는 유전자의 염기 서열 사이의 연관(linkage)을 검정하는 것이다. 이를 통해 임의의 마커에 대해 LD 가 기능을 갖고 있는 유전자와 계속 함께 있는지 를 검정할 수 있는 가능성을 높이는 것이다. 그러나 이 방법 으로 개발된 연관 마커는 근연관계가 없는 옥수수들의 육종 에서 MAS 로 적합하지 않다. 왜냐하면 마커와 유용한 기능 적 변이 사이에서는 연관이 근연관계가 없는 유전자형들의 재조합 과정 도중에 끊어지기 때문이다. 옥수수의 association mapping에 사용되는 임의의 마커는 다양한 association mapping panel에서 LD의 와해(breakdown) 때문에 통계적 유의차가 있는 association을 위한 기능을 갖는 대립유전자에 반드시 매우 가깝게 존재하여야 한다. 많은 세대의 재조합은 두 계 통의 교배에서부터 몇 세대까지 유전적 연관지도 집단 (linkage mapping population)의 계통과 비교되는 가장 최근 의 공통의 조상에서부터 시작된 다양성 association panel에 서 근연관계가 없는 계통을 분리해낸다(Yan et al., 2010). 어떤 주어진 association mapping panel에서 각 개체의 유전 자형 분석에 사용된 마커 가운데서 LD 의 수준은 association 연구를 성공하기 위한 중요한 지표 중 하나인데, 그 이유는 유의차가 있는 연관(association)의 검정을 위해 필요로 하 는 최소한의 마커 수와 해상도를 가늠할 수 있게 도와주기 때문이다(Yan et al., 2009).
Genome-Wide Association Mapping
유전자를 기반으로 한 association mapping은 잠재적 후 보 유전자의 자세한 사전 정보가 요구되는 가정 구동 접근 (hypothesis-driven approach) 방법이다. 옥수수에서 이 연구 방법이 많이 사용되었으나 임의의 마커를 기반으로 한 낮은 association 분석 능력으로 특히 옥수수에서 빠른 LD decay 때문에 문제가 되기도 한다(Gore et al., 2009). 하지만 옥수 수에서 이용할 수 있는 SNP 마커는 점점 증가하여 옥수수 유전체의 모든 염색체에 고루 퍼져있으며, 복잡한 목표 유 전 형질에 가깝게 연관되어 있다. 그래서 GWAS(Genomewide association studies)라고 부르는 연구가 여러 작물에서 폭넓게 이용되고 있다(Altshuler et al., 2008; Hunter and Crawford, 2008). GWAS 는 수십만 개 이상의 SNP를 필요 로 하는 각 LD 블록에서 충분한 다형성을 보이는 마커를 가지고 수집된 각 개체의 유전자형의 특성검정을 가지고 시 작한다. 이 genotyping 의 밀도는 array를 이용한 시스템을 이용하여 동시에 genotyping을 백만 개 씩 할 수 있다 (Gupta et al., 2008; Yan et al., 2010). 그렇지 않으면, 염기 서열 분석 능력의 발달과 NGS 기술을 이용한 단위당 단가 의 하락은 GWA 연구의대용량 genotyping의 필요를 위해 두 개를 연결할 것이다(Metzker et al., 2010).
GWAS를 성공하기 위한 최소한의 필요한 마커의 수는 유전체의 크기와 LD decay 의 비율을 따르게 된다. 예를 들 어 유전체 크기가 125 Mbp인 애기장대의 경우에는 140,000 개의 마커로 충분히 가능하나, 옥수수의 경우는 유전체 크 기가 2300 Mbp 이나 되며 또한 LD decay 비율이 애기장대 보다 훨씬 더 빠르기 때문에 천만 개 이상의 마커가 필요할 것으로 추정하고 있다(Kim et al., 2007; Myles et al., 2009). 그러나 현재로는 실험적인 명확한 마커의 수는 아직 정확히 알 수 없으며, 오직 NGS를 통한 re-sequencing 만이 해당 genotyping의 수준에 대한 마커수를 제공할 수 있으며 그렇기 때문에 reference genome을 가지고 있는 종에서만 가능하다(Lupski et al., 2010). 더욱이 그 결과로 생긴 데이 터들의 통계적 분석은 현재의 데이터 취급의 커다란 도전 과제를 제시하는 것이다. Association mapping panel을 구 축을 위한 엘리트 계통을 이용한다면 옥수수에서 GWAS를 위해 필요한 마커의 수를 대폭 줄일 수 있을 것 이다(Yan et al., 2010). 즉, 모든 유전자의 발현되는 부분을 이용한 다 형성을 통하여 지속적인 QTL 분석의 감소 없이 필요한 마 커의 수를 줄일 수 있다는 의미이다. 이것은 유전자 부분의 다형성이 임의로 선발된 마커를 이용한 다형성과 비교하였 을 때 기능적으로 중요한 것일 것이기 때문이다. 이것은 그 동안 연구된 이미 클로닝 되어 있는 대부분의 QTL에 의해 서도 입증 된다(Alonso-Blanco et al., 2005; Salvi & Tuberosa, 2005). 비록 목표 유전자의 상단과 말달 방향으로 10 kb 정 도의 부분이 빠질 수 있기 때문에 cis- 조절 인자와 같은 기 능적으로 중요한 인자를 놓칠 수 있더라도 단기적으로 GWA는 유전자 중심적 접근을 통하여 발현되는 부분을 중 심으로 진행되게 될 것이다(Jorgenson & Witte, 2006; Ng et al., 2010). 옥수수의 유전체는 이미 염기서열이 밝혀진 B73을 기준으로 32,000 개의 유전자가 있을 것으로 예상하 며 평균적으로 약 1.4 kb의 cDNA 가 생성될 것으로 예상 한다(Alexandrov et al., 2009; Schnable et al., 2009). 그래 서 이런 유전자들의 각 내부의 월등히 우세한 다형성을 찾 아내는 것이 가능할 것이고 GWAS를 위하여 대량의 강력 한 유전자 기반 마커가 제공될 것이다. 만약 옥수수의 유전 자가 5만개이고 각 발현 유전자 부분에서 10∼20개의 마커 가 개발된다고 가정해보면, 만약 50만~100만 개의 잘 선 발된 마커가 이용된다면 옥수수 GWAS는 충분한 QTL 검 정 능력을 가지게 될 것이다. 이렇게 하여 옥수수 유전체의 발현되는 부분으로부터 개발된 마커의 이용은 1000만~ 1500만개가 필요한 것으로 예상하는 임의의 마커를 사용할 때와 비교하여 필요한 마커수를 대략 10~20배로 줄일수 있다(Myles et al., 2009).
앞에서 설명한 NAM 집단은 association mapping을 이용 한 매우 유용한 유전자원이다. NAM 집단은 근본적으로 공 통의 부모에 의해 연관된 25개의 아주 유사한 양친의 집단 의 시리즈이기 때문에 연관 지도(linkage mapping)과 연합 지도(association mapping)의 장점을 모두 가지고 있다. NAM 집단의 디자인에서 가장 뛰어난 장점 중 하나는 정교 한 유전자형이 조상 계통에 대한 정교한 genotyping에 의해 전체 집단에 대해 부여될 수 있다는 것이며, 그래서 자식 계 통에게 저 밀도 유전자 마커를 기반으로 유전자형을 계획할 수 있다는 것이다(Wallace et al., 2014). 이것으로 인하여 GWAS 가 아주 세밀한 규모에서 형질과 유전자 좌 사이의 association을 찾기 위한 조상집단의 재조합 과정에 대한 모 든 장점을 가질 수 있게 한다(Fig. 1(B)). NAM 집단 외에도 많은 정교한 association 집단이 GWAS와 연계되고 있다 (Fint-Garcia et al., 2005; Yang et al., 2010).
결 론
DNA를 기반으로 한 분자 마커가 처음 개발된 지 30년이 넘었다. 그 이후로 많은 연구자들은 수많은 분자마커를 개 발하고, 직접 활용하기 위한 이용성을 증명하기 위하여 많 은 노력을 기울였다. 최근 들어 옥수수에서 MAS를 활용하 기 위한 기술적 기회는 급격히 증가하고 있으며, 그로인해 국제적으로 옥수수 유전과 육종에 대한 생물 다양성을 위한 분자 마커의 이용성이 활발히 연구 되고 있으나 여전히 가 야할 길은 멀다. 옥수수 germplasm에서 표현형과 분자 마 커를 이용한 다양성 검정과 특히 한발 스트레스나 노균병에 대한 저항성과 같은 다양한 농업 형질에 영향을 미치는 QTL의 발굴 등이 많은 나라에서 발 빠르게 진행되고 있다. 그러나 마커를 육종에 응용하여 옥수수 종실의 생산성을 증 가시키는 연구는 우리나라는 물론 동남아시아 일대에서는 여전히 취약한 분야이다. 이를 해결하기 위해서는 분자마커 를 육종에 활용하는 것은 매우 유용한 방법으로 평가 받는 다. 과거에는 작물 육종가들은 자식계통과 표현형 분석, 일 반적이고 특별하게 조합된 능력의 지식이 포함된 데이터베 이스를 이용하여 육종을 하였으나 이제는 작물 육종가가 분 자 마커와 지문법, 마커와 표현형이 연관된 작물 유전체 전 체를 이용하는 데이터베이스를 이용하는 추세로 변하고 있 다. 또한 분자마커기술을 이용한 다양성 분석이 폭넓게 이 루어지고 있으며, 대량 변이 분석을 위한 DNA chip 기술과 database를 이용한 생명정보 기술은 앞으로 더욱 중요하게 이용될 것이다. 마지막으로 분자 육종가들이 분자마커를 육 종에 활용하기 위한 기술 개발에 더욱 현명하고 더욱 집중 적인 실질적인 해결방안을 개발하기 위하여 더욱 노력해야 할 것이다.
